Unternehmen, die an den Grenzen der Luft- und Raumfahrt, Energie und Informatik arbeiten, sind ständig auf der Suche nach neuen Materialien, um die Leistung zu verbessern. Doch um zu verstehen, wie sich diese Materialien tatsächlich verhalten, wenn sie in Raketen oder auf Computerchips eingesetzt werden, müssen Unternehmen das Materials zunächst herstellen und dann testen. Das liegt daran, dass selbst die leistungsstärksten Simulationstechniken Schwierigkeiten haben, die komplexen chemischen Anordnungen in den meisten heutigen Feststoffmaterialien zu modellieren. Das Downside erhöht die Kosten und den Zeitaufwand für die Materialinnovation.
Jetzt hat ein Staff von MIT-Forschern eine Möglichkeit geschaffen, das Verhalten von Metallen unabhängig von der Komplexität ihrer chemischen Anordnung genau zu modellieren. Im Zentrum des Ansatzes stehen maschinelle Lernmodelle, die Materialsimulationen schneller und genauer machen. Die Forscher verbesserten diese Modelle, indem sie Trainingsdatensätze erstellten, die die Vielfalt atomarer Umgebungen in chemisch ungeordneten Materialien erfassen.
In einem neues Papier rein Fortschritte in den Wissenschaftenzeigten die Forscher, dass ihr Ansatz zur genauen Vorhersage von Materialeigenschaften für eine vielfältige Gruppe von Metalllegierungen unter verschiedenen Bedingungen verwendet werden kann. Sie zeigten auch, wie der Ansatz zur Entwicklung neuer Materialien genutzt werden könnte, insbesondere in Szenarien, in denen Experimente teuer sind.
„Der Schwerpunkt der Arbeit liegt auf metallischen Legierungen, dem Fachgebiet, in dem ich arbeite, aber dies könnte auf andere Arten von Materialien, wie etwa Halbleiter, übertragen werden“, sagt der leitende Autor Rodrigo Freitas, TDK Profession Improvement Professor für Materialwissenschaft und Werkstofftechnik am MIT. „Das ist nicht spezifisch für eine bestimmte Anwendung – Sie könnten diesen Ansatz nutzen, um neue nachhaltige Stähle, neue Materialien für die Luft- und Raumfahrt und mehr zu entwickeln. Das macht das Spannende.“
Zu Freitas gesellen sich bei der Arbeit der Erstautor Killian Sheriff PhD ’26; MIT-Doktoranden Daniel Xiao und Yifan Cao; und Lewis R. Owen, Dozent an der College of Sheffield.
Modellieren von Metallen
Materialeigenschaften werden größtenteils durch die innere Anordnung ihrer chemischen Elemente bestimmt. Selbst wenn zwei Materialien die gleiche Mischung chemischer Elemente aufweisen, können unterschiedliche chemische Anordnungen den Unterschied zwischen einem spröden Materials und einem Materials, das sich verformt, ohne zu brechen, ausmachen.
Um diesen Unterschied zu erfassen, müssen Materialien Atom für Atom simuliert werden. Dazu stützen sich Forscher auf Modelle, die beschreiben, wie Atome miteinander interagieren. In den letzten zwei Jahrzehnten hat sich maschinelles Lernen zur genauesten Methode zur Erstellung dieser Modelle entwickelt. Solche Modelle funktionieren intestine, wenn die chemischen Anordnungen innerhalb von Materialien streng geordneten Mustern folgen. Dies ist jedoch bei den meisten festen Materialien nicht der Fall, deren atomare chemische Anordnungen ungeordnet sind und von Area zu Area variieren.
„Die eigentliche Herausforderung auf unserem Gebiet besteht darin, diese chemisch ungeordneten Phasen zu modellieren“, sagt Freitas. „Chemische Unordnung bedeutet, dass es eine große Vielfalt lokaler chemischer Umgebungen gibt, die für das maschinelle Lernmodell schwer zu erlernen sind. Das ist ein Downside, weil jedes einzelne Metall, das wir in der Praxis verwenden, chemisch ungeordnet ist.“
Das Downside beruht auf dem Mangel an repräsentativen Trainingsdaten für diese Atom-für-Atom-Simulationen. Der derzeit führende Ansatz zur Erstellung solcher Daten funktioniert mit roher Gewalt und erfordert oft mehr als 100.000 Rechenstunden, um die Trainingsdaten für ein einzelnes Materials zu erstellen. Selbst dann lässt es sich nicht intestine übertragen, wenn Forscher die Zusammensetzung des Supplies ändern.
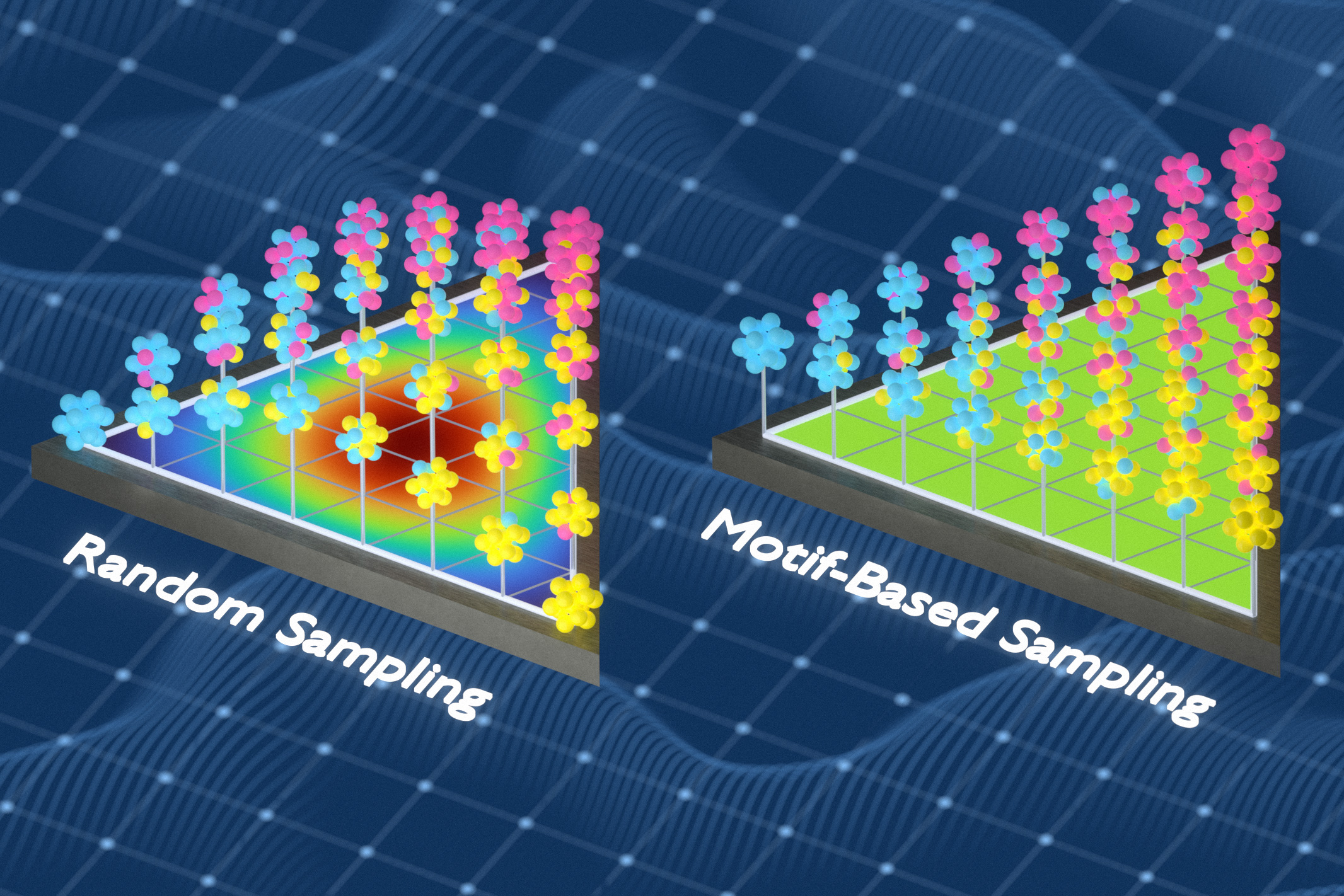
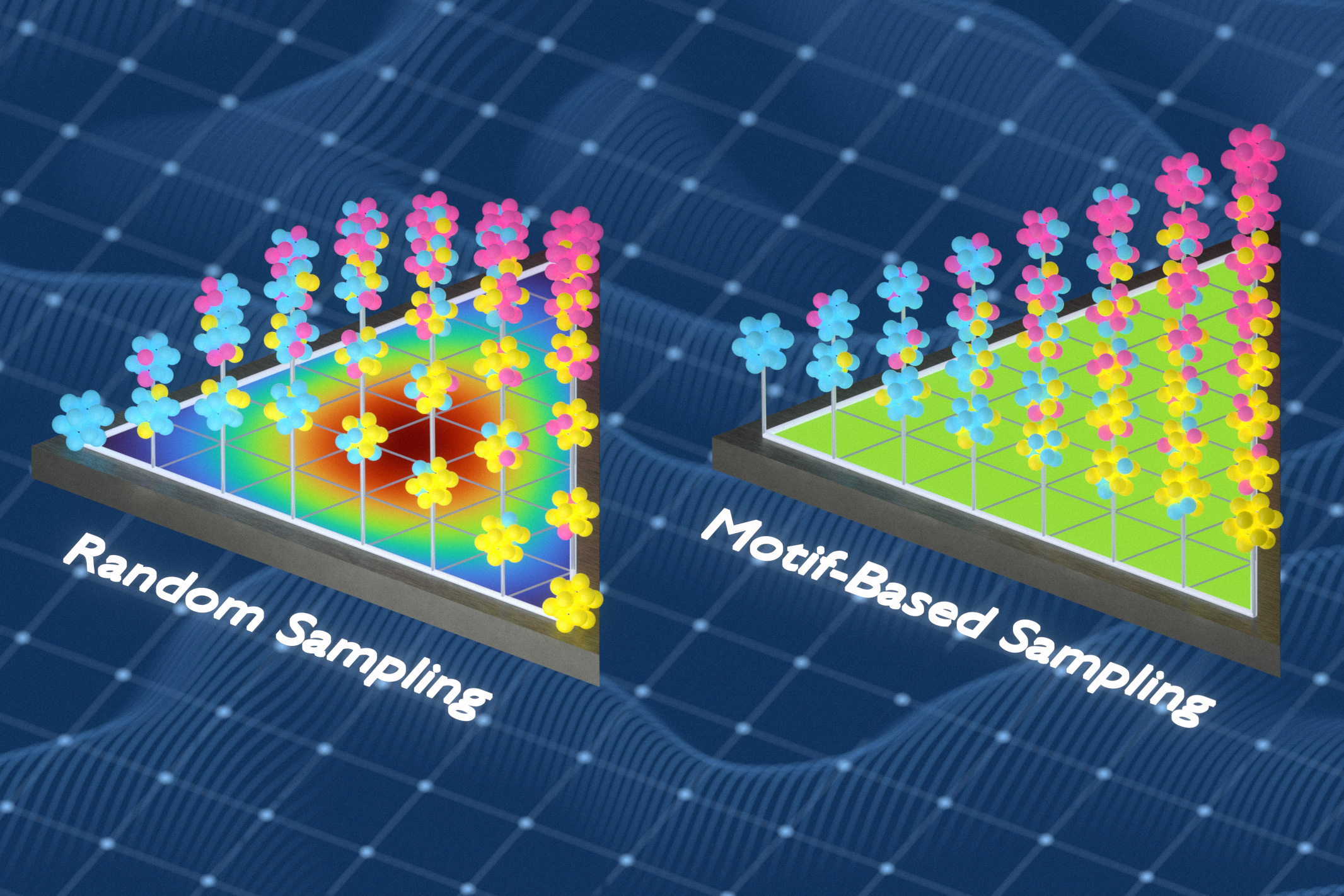
In bisherige ArbeitFreitas‘ Gruppe hatte eine Methode entwickelt, um die chemische Komplexität fester Materialien zu messen, indem sie die Häufigkeit und den Abstand winziger Atomgruppen analysierte. Für diese Studie nutzten die Forscher diese Fähigkeit, um bessere Trainingsdatensätze zu erstellen. Sie verwendeten einen mathematischen Ansatz, der als Informationstheorie bekannt ist, um Trainingsdatensätze zu generieren, die eine größere Vielfalt lokaler chemischer Umgebungen in ungeordneten Materialien erfassen. Bei der Methode werden Atome aus Proben ausgetauscht, um Wiederholungen zu reduzieren und das Modell chemischen Umgebungen auszusetzen, die es andernfalls übersehen würde.
„Wir haben das Trainingsset ständig optimiert, damit es so viele verschiedene lokale Umgebungen wie möglich erfasst“, sagt Freitas. „Wenn die gleiche Artwork von Umgebung viele Male auftauchte, ersetzten wir redundante Beispiele durch solche, die das Modell zuvor nicht gesehen hatte. Das macht den Trainingssatz viel informativer, weil jedes Beispiel etwas Neues hinzufügt.“
Beim Coaching anhand der Datensätze der Forscher sagten die Modelle Materialeigenschaften genauer voraus als Modelle, die mithilfe von Zufallsstichproben oder einer anderen gängigen Stichprobenmethode trainiert wurden.
„Der Ausgangspunkt all dieser Atom-für-Atom-Simulationen ist: Können Sie die chemische Bindung zwischen Atomen genau beschreiben?“ Freitas erklärt. „Wenn nicht, können Sie damit immer noch etwas über Materialien im Allgemeinen lernen, aber es sagt Ihnen nicht, was mit bestimmten Materialien in der realen Welt passieren wird. Dieser Ansatz sorgt dafür, dass die Simulationen hinsichtlich ihrer Chemie eine hohe Wiedergabetreue erhalten, um besser widerzuspiegeln, was mit Materialien geschieht.“
Die Forscher wandten ihre Technik an, um Trainingsdatensätze für maschinelles Lernen für eine Gruppe chemisch unterschiedlicher Metalllegierungen zu erstellen. Mithilfe einer Reihe von Modellen für maschinelles Lernen zeigten sie, dass die auf ihren Datensätzen trainierten Modelle genauer sind als viel größere Modelle, die von Unternehmen wie Google und Microsoft erstellt wurden.
„Wir waren an einem Punkt angelangt, an dem wir überzeugt waren, dass es ohne den Einsatz dieser teuren Brute-Power-Methoden funktioniert“, sagt Freitas. „Ich sagte zu Killian: ‚Das ist eine gute Arbeit. Aber wenn man zeigen kann, dass Simulationen mit diesen Modellen nun nützliche Materialeigenschaften genau vorhersagen können, dann wird es eine sehr gute Arbeit.‘ Killian hat sich das zu Herzen genommen und es so umfassend wie möglich getestet.“
Sheriff arbeitete mit Xiao und Cao zusammen, um den Ansatz für verschiedene Legierungen und Eigenschaften zu testen. Das Staff nutzte auch Owens experimentelle Daten, um die Simulationen mit realen Messungen der atomaren Ordnung in Legierungen zu vergleichen.
Vom Labor in die Industrie
Die Methode funktioniert zum Teil durch die Erfassung verborgener Muster in den Beispieldaten. Die Forscher beschreiben die Muster in der Arbeit als „subtile energetische Tendenzen hin zu bestimmten lokalen chemischen Konfigurationen“.
Diese kleinen energetischen Unterschiede sind wichtig, weil sie bestimmen, welche Phasen sich in einer Legierung bilden, wie sich diese Phasen mit der Temperatur und der Zusammensetzung ändern und letztendlich welche Eigenschaften das Materials haben wird. Als einen Check führte Daniel Xiao Simulationen durch, die zeigten, dass die Modelle des Groups Phasendiagramme vorhersagen konnten, die den experimentellen Daten weitgehend entsprachen. Phasendiagramme zeigen, welche Phasen bei verschiedenen Temperaturen und chemischen Zusammensetzungen stabil sind, und sind ein zentrales Werkzeug für die Konstruktion und Verarbeitung von Legierungen.
„Phasendiagramme sind eine der wichtigsten Methoden, mit denen Menschen die Materialmodellierung mit realen Verarbeitungsentscheidungen verbinden“, sagt Freitas. „Wenn Sie eine Legierung schweißen, gießen oder wärmebehandeln, müssen Sie wissen, welche Phasen sich unter verschiedenen Bedingungen wahrscheinlich bilden. Unser Ziel ist es, diese Artwork von Vorhersagen so genau und zugänglich zu machen, dass sie Teil der Artwork und Weise werden, wie Menschen Materialien entwerfen.“
Die Forscher verwenden diesen Ansatz nun, um zu untersuchen, wie sich eine Änderung der Zusammensetzung einer Legierung auf die mechanischen Eigenschaften und die Strahlungstoleranz auswirkt, mit dem Ziel, Materialien zu entwickeln, die auch in rauen Umgebungen stark und schadenstolerant bleiben. Sie arbeiten auch daran, die Anwendung der Methode mit den Arten von Werkzeugen und Arbeitsabläufen zu vereinfachen, auf die sich Materialingenieure bereits verlassen.
„Die Industrie wird ihre Arbeitsweise nicht ändern, wenn das, was Sie erstellen, nicht in ihre bestehenden Betriebsabläufe passt“, sagt Freitas. „Ziel ist es, diese Vorhersagen dort nutzbar zu machen, wo tatsächlich Materialentscheidungen getroffen werden.“
Die Forschung wurde vom US Air Power Workplace of Scientific Analysis unterstützt.