
Alle biologischen Funktionen hängen davon ab, wie unterschiedliche Proteine miteinander interagieren. Protein-Protein-Wechselwirkungen erleichtern alles von der transkribierenden DNA und der Kontrolle der Zellteilung bis hin zu Funktionen auf höherer Ebene in komplexen Organismen.
Es bleibt jedoch viel unklar darüber, wie diese Funktionen auf molekularer Ebene orchestriert werden und wie Proteine miteinander interagieren – entweder mit anderen Proteinen oder mit Kopien von sich.
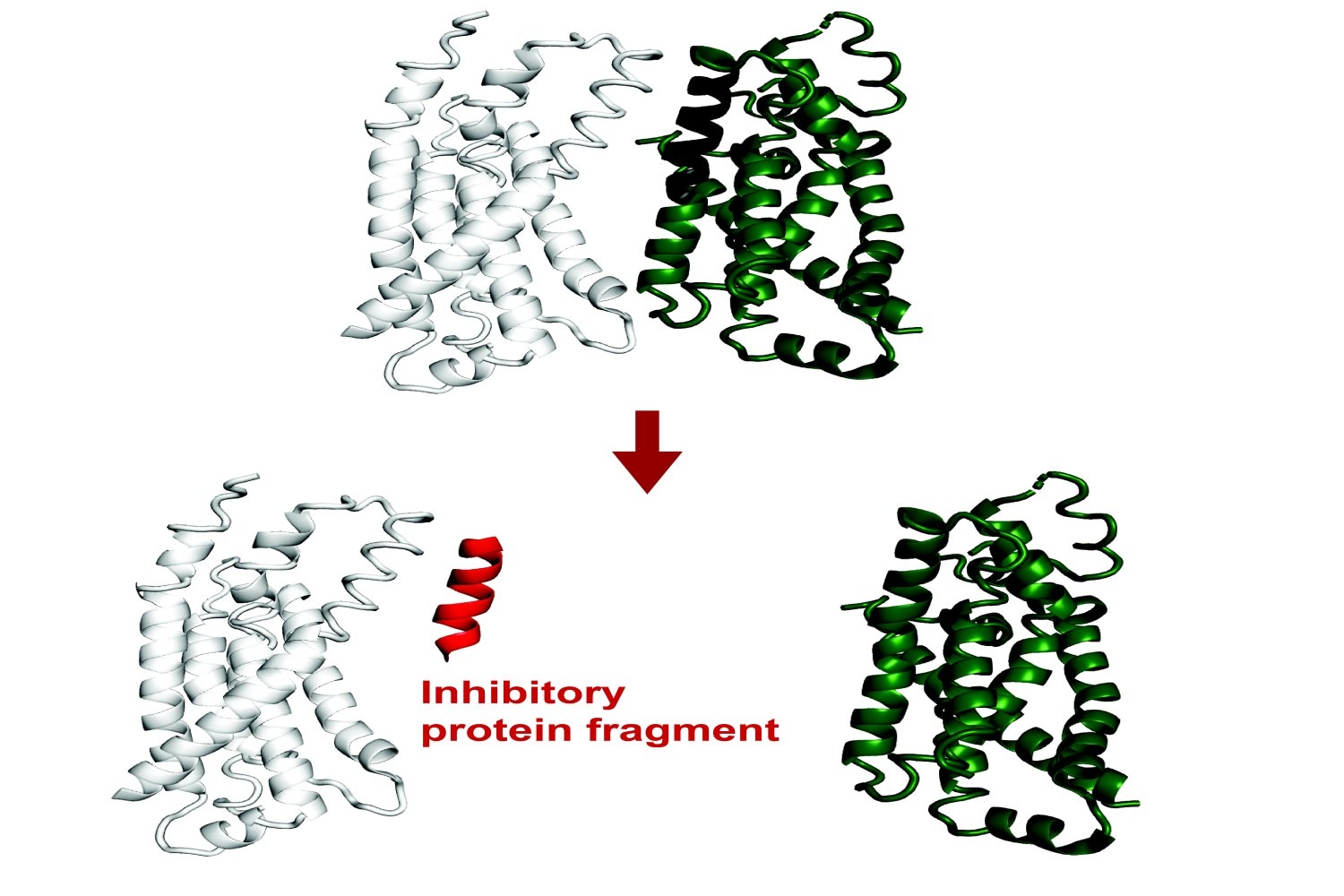
Jüngste Erkenntnisse haben gezeigt, dass kleine Proteinfragmente ein großes funktionelles Potenzial haben. Obwohl es sich um unvollständige Stücke handelt, können kurze Aminosäurenabschnitte immer noch an Grenzflächen eines Zielproteins binden, was native Wechselwirkungen rekapituliert. Durch diesen Prozess können sie die Funktion dieses Proteins ändern oder seine Wechselwirkungen mit anderen Proteinen stören.
Proteinfragmente könnten daher sowohl die Grundlagenforschung zu Proteinwechselwirkungen als auch zelluläre Prozesse ermöglichen und möglicherweise therapeutische Anwendungen aufweisen.
Kürzlich veröffentlicht in Verfahren der Nationalen Akademie der Wissenschafteneine neue Methode, die im Division of Biology entwickelt wurde E. coli. Theoretisch könnte dieses Instrument zu genetisch kodbaren Inhibitoren gegen jedes Protein führen.
Die Arbeit wurde im Labor des außerordentlichen Professors für Biologie und im Forscher des Howard Hughes Medical Institute erledigt Gene-Wei li In Zusammenarbeit mit dem Labor von Jay A. Stein (1968) Professor für Biologie, Professor für Biologische Ingenieurwesen und Abteilungsleiter Amy Keating.
Nutzung des maschinellen Lernens
Das Programm namens Fragfold nutzt Alphafold, ein KI -Modell, das in den letzten Jahren zu phänomenalen Fortschritten in der Biologie geführt hat, da die Proteinfaltung und Proteinwechselwirkungen vorhergesagt werden können.
Ziel des Projekts struggle es, Fragmentinhibitoren vorherzusagen, was eine neuartige Anwendung von Alphafold ist. Die Forscher dieses Projekts bestätigten experimentell, dass mehr als die Hälfte der Vorhersagen von Fragfold für die Bindung oder Hemmung genau waren, selbst wenn die Forscher keine früheren strukturellen Daten zu den Mechanismen dieser Wechselwirkungen hatten.
„Unsere Ergebnisse legen nahe, dass dies ein verallgemeinerbarer Ansatz ist, um Bindungsmodi zu finden, die wahrscheinlich die Proteinfunktion hemmen, einschließlich neuer Proteinziele, und Sie können diese Vorhersagen als Ausgangspunkt für weitere Experimente verwenden“, sagt Co-First und entsprechender Autor Andrew Savinov, ein Postdoc im Li -Labor. „Wir können dies wirklich auf Proteine ohne bekannte Funktionen ohne bekannte Interaktionen ohne bekannte Strukturen anwenden, und wir können diese Modelle, die wir entwickeln, etwas Glaubwürdigkeit einbringen.“
Ein Beispiel ist FTSZ, ein Protein, das für die Zellteilung von entscheidender Bedeutung ist. Es ist intestine untersucht, enthält aber eine Area, die intrinsisch ungeordnet ist und daher besonders herausfordernd zu studieren ist. Störte Proteine sind dynamisch, und ihre funktionellen Wechselwirkungen sind sehr wahrscheinlich flüchtig – so kurz, dass aktuelle Strukturbiologie -Instruments keine einzelne Struktur oder Interaktion erfassen können.
Die Forscher nutzten Fragfold, um die Aktivität von Fragmenten von FTSZ, einschließlich Fragmenten der intrinsisch ungeordneten Area, zu untersuchen, um mehrere neue Bindungswechselwirkungen mit verschiedenen Proteinen zu identifizieren. Dieser Sprung in das Verständnis bestätigt und erweitert frühere Experimente zur missischen biologischen Aktivität von FTSZ.
Dieser Fortschritt ist teilweise von Bedeutung, weil er ohne die Lösung der Struktur der ungeordneten Area durchgeführt wurde, und weil sie die potenzielle Kraft von Fragfold aufweist.
„Dies ist ein Beispiel dafür, wie Alphafold sich grundlegend verändert, wie wir Molekül- und Zellbiologie untersuchen können“, sagt Keating. „Kreative Anwendungen von KI -Methoden, wie unsere Arbeit an Fragfold, eröffnen unerwartete Fähigkeiten und neue Forschungsrichtungen.“
Hemmung und darüber hinaus
Die Forscher erfüllten diese Vorhersagen, indem sie jedes Protein rechnerisch fragmentiert und dann modellierten, wie diese Fragmente an Interaktionspartner binden würden, die sie für related hielten.
Sie verglichen die Karten der vorhergesagten Bindung über die gesamte Sequenz mit den Effekten derselben Fragmente in lebenden Zellen, die unter Verwendung experimenteller Messungen mit hohem Durchsatz bestimmt wurden, bei denen Millionen von Zellen jeweils eine Artwork Proteinfragment produzieren.
Alphafold verwendet koevolutionäre Informationen, um die Faltung vorherzusagen, und bewertet typischerweise die Evolutionsgeschichte von Proteinen anhand von etwas, das für jeden einzelnen Vorhersagelauf als mehrere Sequenzausrichtungen bezeichnet wird. Die MSAs sind kritisch, sind jedoch ein Engpass für groß angelegte Vorhersagen-sie können eine unerschwingliche Zeit und Rechenleistung erfordern.
Für Fragfold präkulierten die Forscher stattdessen einmal die MSA für ein Protein in voller Länge und verwendeten dieses Ergebnis, um die Vorhersagen für jedes Fragment dieses Proteins in voller Länge zu leiten.
Savinov prognostizierte zusammen mit dem Keating Lab -Alumnus Sebastian Swanson, PhD ’23, zusätzlich zu FTSZ inhibitorische Fragmente einer Vielzahl von Proteinen. Zu den Wechselwirkungen gehörte ein Komplex zwischen Lipopolysaccharid -Transportproteinen LPTF und LPTG. Ein Proteinfragment von LPTG inhibierte diese Wechselwirkung und stört vermutlich die Abgabe von Lipopolysaccharid, was eine entscheidende Komponente der ist E. coli Außenzellmembran für die zelluläre Health.
„Die große Überraschung struggle, dass wir die Bindung mit einer solchen hohen Genauigkeit vorhersagen können und die Bindung, die der Hemmung entspricht, häufig voraussagen können“, sagt Savinov. „Für jedes Protein, das wir uns angesehen haben, konnten wir Inhibitoren finden.“
Die Forscher konzentrierten sich zunächst auf Proteinfragmente als Inhibitoren, denn ob ein Fragment eine wesentliche Funktion in Zellen blockieren könnte, ist ein relativ einfaches Ergebnis, um systematisch zu messen. Mit Blick nach vorne ist Savinov auch daran interessiert, die Fragmentfunktion außerhalb der Hemmung zu untersuchen, wie Fragmente, die das Protein stabilisieren können, an das sie binden, ihre Funktion verbessern oder verändern oder den Proteinabbau auslösen.
Design im Prinzip
Diese Forschung ist ein Ausgangspunkt für die Entwicklung eines systemischen Verständnisses von Prinzipien für zelluläre Designs und die Elemente, auf die Tiefe-Studying-Modelle abrufen können, um genaue Vorhersagen zu treffen.
„Es gibt ein breiteres, weiter reichhaltiges Ziel, das wir aufbauen“, sagt Savinov. „Jetzt, da wir sie vorhersagen können, können wir die Daten verwenden, die wir aus Vorhersagen und Experimenten haben, um die herausragenden Merkmale herauszuholen, um herauszufinden, was Alphafold tatsächlich gelernt hat, was einen guten Inhibitor ausmacht?“
Savinov und Mitarbeiter haben sich auch weiter mit der Bindung von Proteinfragmenten befassten, um andere Proteinwechselwirkungen zu untersuchen und spezifische Reste zu mutieren, um zu sehen, wie diese Wechselwirkungen die Artwork und Weise verändern, wie das Fragment mit seinem Ziel interagiert.
Experimentell untersuchte das Verhalten von Tausenden von mutierten Fragmenten innerhalb von Zellen, ein Ansatz, der als Deep Mutational Scanning bekannt ist, Schlüsselaminosäuren, die für die Hemmung verantwortlich sind. In einigen Fällen waren die mutierten Fragmente noch stärker inhibitoren als ihre natürlichen Sequenzen in voller Länge.
„Im Gegensatz zu früheren Methoden sind wir nicht darauf beschränkt, Fragmente in experimentellen Strukturdaten zu identifizieren“, sagt Swanson. „Die Kernstärke dieser Arbeit ist das Zusammenspiel zwischen experimentellen Hemmungdaten mit hohem Durchsatz und den vorhergesagten Strukturmodellen: Die experimentellen Daten führen uns zu den besonders interessanten Fragmenten, während die von Fragfold vorhergesagten Strukturmodelle eine spezifische, Testbare Hypothese für liefern Wie die Fragmente auf molekularer Ebene funktionieren. “
Savinov freut sich über die Zukunft dieses Ansatzes und seine unzähligen Anwendungen.
„Durch die Erzeugung kompakter, genetisch kodierbarer Bindemittel eröffnet Fragfold eine breite Palette von Möglichkeiten zur Manipulation der Proteinfunktion“, stimmt Li zu. „Wir können uns vorstellen, funktionalisierte Fragmente zu liefern, die native Proteine modifizieren, ihre subzelluläre Lokalisierung ändern und sogar neu programmieren, um neue Werkzeuge für die Untersuchung der Zellbiologie und die Behandlung von Krankheiten zu erstellen.“