Der Prozess der Entdeckung von Molekülen, die über die Eigenschaften verfügen, die zur Schaffung neuer Medikamente und Materialien erforderlich sind, ist umständlich und teurer, umfangreicher Rechenressourcen und Monate menschlicher Arbeitskräfte, um den enormen Raum potenzieller Kandidaten einzugrenzen.
Großsprachige Modelle (LLMs) wie ChatGPT könnten diesen Prozess rationalisieren, aber es ermöglicht, einen LLM zu verstehen und zu begründen, dass die Atome und Bindungen, die ein Molekül bilden, genauso wie mit Wörtern, die Sätze bilden, einen wissenschaftlichen Stolperstein dargestellt haben.
Forscher von MIT und dem MIT-IBM Watson AI Lab haben einen vielversprechenden Ansatz erstellt, der ein LLM mit anderen maschinell-lernenden Modellen erweitert, die als graphbasierte Modelle bekannt sind und speziell zur Erzeugung und Vorhersage von Molekülstrukturen entwickelt wurden.
Ihre Methode verwendet eine Foundation -LLM, um Abfragen der natürlichen Sprache zu interpretieren, die die gewünschten molekularen Eigenschaften angeben. Es wechselt automatisch zwischen den Basisllm- und graphbasierten KI-Modulen, um das Molekül zu entwerfen, die Begründung zu erklären und einen Schritt-für-Schritt-Plan zu erzeugen, um es zu synthetisieren. Es verschachtelt Textual content-, Graph- und Synthese -Schrittgenerierung, kombiniert Wörter, Diagramme und Reaktionen in ein gemeinsames Vokabular, damit das LLM konsumiert werden kann.
Im Vergleich zu vorhandenen LLM-basierten Ansätzen erzeugte diese multimodale Technik Moleküle, die die Benutzerspezifikationen besser übereinstimmten und mit größerer Wahrscheinlichkeit einen gültigen Synthesisplan aufweisen, was das Erfolgsverhältnis von 5 Prozent auf 35 Prozent verbesserte.
Es übertraf auch LLMs, die mehr als das 10-fache der Größe haben und die Moleküle und Synthese-Routen nur mit textbasierten Darstellungen entwerfen, was darauf hindeutet, dass Multimodalität der Schlüssel zum Erfolg des neuen Programs ist.
„Dies könnte hoffentlich eine Finish-to-Finish-Lösung sein, bei der wir von Anfang bis Ende den gesamten Prozess des Entwerfens und Herstellens eines Moleküls automatisieren würden. Wenn ein LLM nur die Antwort in wenigen Sekunden geben könnte, wäre dies ein großer Zeitspur für Pharmaunternehmen“, sagt Michael Solar, ein MIT-Absolventenstudent und der Co-Autor eines MIST-Absolventen und Co-Autor eines AIT-Autors eines A-Autors eines A-Autors eines Eins eines Eins eines Eins eines Eins eines MET-Unternehmens eines MET-Absolventen eines MET-Absolventen eines Einsatzes eines Einsatzes eines Eins eines Eins eines Eins eines MET-Unternehmens eines MET-Absolventen eines MET-Unternehmens eines MET-Unternehmens wäre ein großer Zeitspur für einen Einsatz eines Pharmazeutikums einer LLM. Papier über diese Technik.
Zu den Mitautoren von Solar gehören die Hauptautorin Gang Liu, eine Doktorandin an der Universität von Notre Dame; Wojciech Matusik, Professor für Elektrotechnik und Informatik am MIT, der die Rechendesign- und Fabricationsgruppe innerhalb des Labors für Informatik und künstliche Intelligenz (CSAIL) leitet; Meng Jiang, außerordentlicher Professor an der Universität von Notre Dame; und Senior-Autorin Jie Chen, leitender Forschungswissenschaftler und Managerin im MIT-IBM Watson AI Lab. Die Forschung wird auf der Internationalen Konferenz über Lernrepräsentationen vorgestellt.
Beste aus beiden Welten
Großsprachenmodelle werden nicht gebaut, um die Nuancen der Chemie zu verstehen. Dies ist ein Grund, warum sie mit inverser molekularer Design zu kämpfen haben, ein Prozess der Identifizierung molekularer Strukturen, die bestimmte Funktionen oder Eigenschaften haben.
LLMs konvertieren Textual content in Darstellungen, die als Token bezeichnet werden und die sie verwenden, um das nächste Wort in einem Satz nacheinander vorherzusagen. Moleküle sind jedoch „Grafikstrukturen“, die aus Atomen und Bindungen ohne bestimmte Reihenfolge bestehen, wodurch es schwierig ist, sie als sequentielle Textual content zu codieren.
Andererseits repräsentieren leistungsstarke KI-Modelle basierte graphbasierte AI-Modelle Atome und molekulare Bindungen als miteinander verbundene Knoten und Kanten in einem Diagramm. Obwohl diese Modelle für inverse molekulares Design beliebt sind, erfordern sie komplexe Eingaben, können die natürliche Sprache nicht verstehen und Ergebnisse erzielen, die schwer zu interpretieren sein können.
Die MIT-Forscher kombinierten ein LLM mit graphbasierten KI-Modellen in einem einheitlichen Rahmen, der das Beste aus beiden Welten holt.
Lamol, das für ein großes Sprachmodell für die molekulare Entdeckung steht, verwendet eine Foundation-LLM als Gatekeeper, um die Abfrage eines Benutzers zu verstehen-eine einfache Sprachanfrage für ein Molekül mit bestimmten Eigenschaften.
Zum Beispiel sucht ein Benutzer möglicherweise ein Molekül, das in die Blut-Hirn-Schranke eindringen und HIV hemmen kann, da es ein Molekulargewicht von 209 und bestimmte Bindungseigenschaften aufweist.
Da der LLM Textual content als Antwort auf die Abfrage vorhersagt, wechselt sie zwischen den Grafikmodulen.
Ein Modul verwendet ein Graph -Diffusionsmodell, um die molekulare Struktur zu erzeugen, die auf Eingangsanforderungen konditioniert ist. Ein zweites Modul verwendet ein Diagramm neuronales Netzwerk, um die erzeugte molekulare Struktur wieder in Token zu codieren, damit die LLMs konsumiert werden können. Das endgültige Diagrammmodul ist ein Diagrammreaktionsprädiktor, der als Eingabe eine Zwischenmolekülstruktur annimmt und einen Reaktionsschritt vorhersagt, der nach den genauen Satz von Schritten sucht, um das Molekül aus grundlegenden Bausteinen herzustellen.
Die Forscher haben eine neue Artwork von Set off -Token erstellt, die dem LLM mitteilt, wann jedes Modul aktiviert werden soll. Wenn das LLM ein Set off -Token „Design“ vorhersagt, wechselt es zu dem Modul, das eine molekulare Struktur skizziert, und wenn es ein „Retro“ -Strigger -Token vorhersagt, wechselt es zu dem retrosynthetischen Planungsmodul, das den nächsten Reaktionschritt vorhersagt.
„Das Schöne daran ist, dass alles, was das LLM erzeugt, bevor ein bestimmtes Modul aktiviert wird, in dieses Modul selbst eingespeist wird. Das Modul lernt, auf eine Weise zu arbeiten, die mit dem übereinstimmt, was zuvor gekommen ist“, sagt Solar.
Auf die gleiche Weise wird die Ausgabe jedes Moduls codiert und wieder in den Erzeugungsprozess des LLM eingespeist. Daher versteht es, was jedes Modul getan hat, und wird weiterhin auf diesen Daten prognostizieren.
Bessere, einfachere molekulare Strukturen
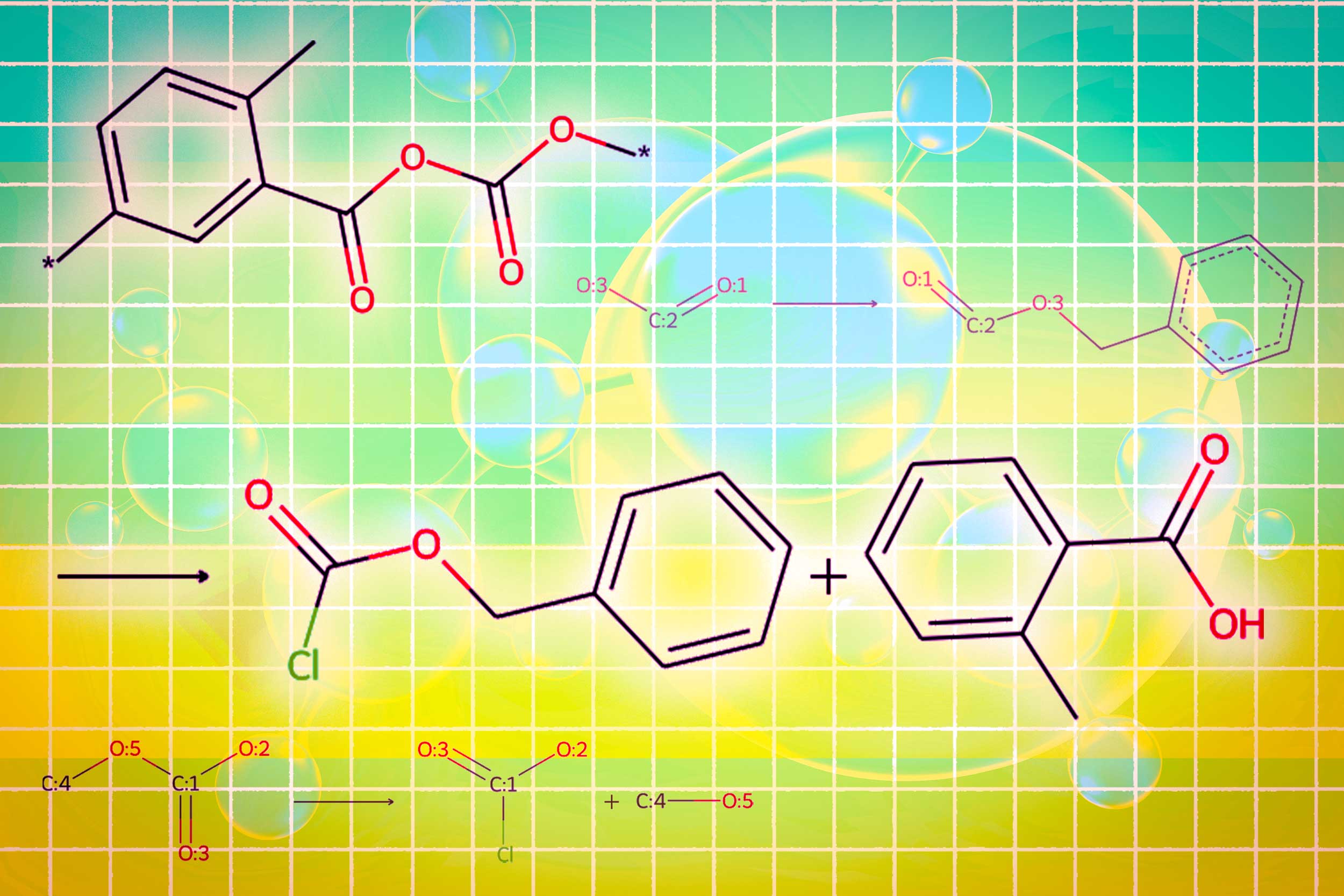
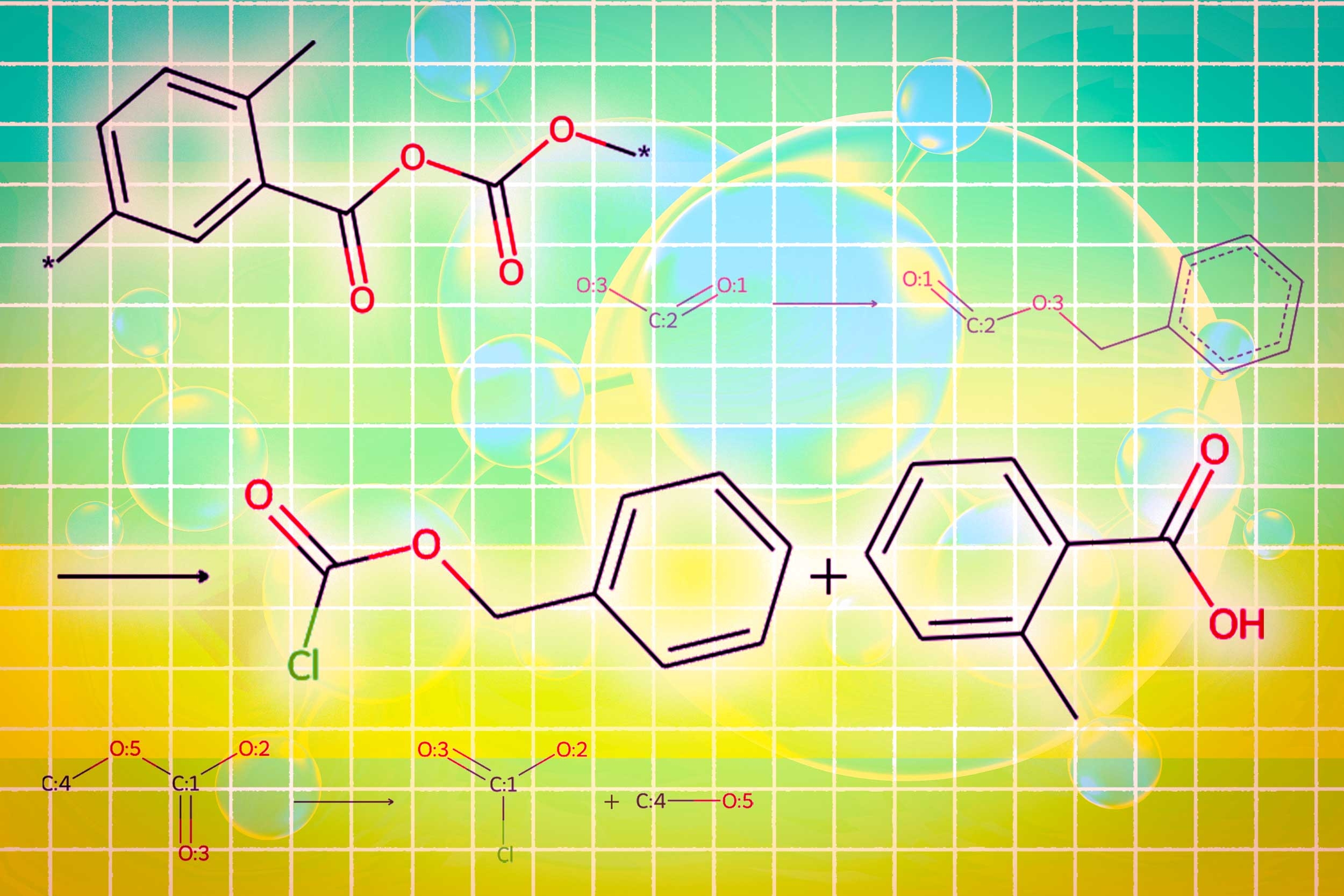
Am Ende gibt Lamol ein Bild der molekularen Struktur, eine Textbeschreibung des Moleküls und einen Schritt-für-Schritt-Syntheseplan aus, der die Einzelheiten zur Herstellung von individuellen chemischen Reaktionen enthält.
In Experimenten mit dem Entwerfen von Molekülen, die die Benutzerspezifikationen entsprechen, übertraf Llamol 10 Commonplace-LLMs, vier fein abgestimmte LLMs und eine hochmoderne domänenspezifische Methode. Gleichzeitig erhöhte es die Erfolgsrate für retrosynthetische Planung von 5 Prozent auf 35 Prozent, indem sie Moleküle erzeugten, die höherwertig sind, was bedeutet, dass sie einfachere Strukturen und kostengünstigere Bausteine hatten.
„LLMs haben Schwierigkeiten, Moleküle zu synthetisieren, da es eine Menge mehrstufiger Planung erfordert. Unsere Methode kann bessere molekulare Strukturen erzeugen, die auch einfacher zu synthetisieren sind“, sagt Liu.
Um Lamol auszubilden und zu bewerten, bauten die Forscher zwei Datensätze von Grund auf neu, da vorhandene Datensätze mit molekularen Strukturen nicht genügend Particulars enthielten. Sie erweiterten Hunderttausende patentierter Moleküle mit mit A-generierten natürlichen Sprachbeschreibungen und maßgeschneiderten Beschreibungsvorlagen.
Der Datensatz, den sie zur Feinabstimmung des LLM erstellt haben, enthält Vorlagen, die sich auf 10 molekulare Eigenschaften beziehen. Eine Lamolbeschränkung besteht darin, dass es geschult ist, Moleküle unter Berücksichtigung der 10 numerischen Eigenschaften zu entwerfen.
In zukünftigen Arbeiten möchten die Forscher Lamol verallgemeinern, damit sie jede molekulare Eigenschaft einbeziehen kann. Darüber hinaus planen sie, die Graphenmodule zu verbessern, um die Erfolgsrate von Lamol von Lamol zu steigern.
Langfristig hoffen sie, diesen Ansatz zu verwenden, um über Moleküle hinauszugehen und multimodale LLMs zu erstellen, die andere Arten von draphbasierten Daten verarbeiten können, z. B. miteinander verbundene Sensoren in einem Stromnetz oder Transaktionen in einem Finanzmarkt.
„Lamol demonstriert die Machbarkeit der Verwendung von großsprachigen Modellen als Schnittstelle zu komplexen Daten, die über die textliche Beschreibung hinausgehen, und wir erwarten, dass sie eine Grundlage sein, die mit anderen AI -Algorithmen interagiert, um alle Diagrammprobleme zu lösen“, sagt Chen.
Diese Forschung wird zum Teil vom MIT-IBM Watson AI Lab, der Nationwide Science Basis und des Workplace of Naval Analysis finanziert.