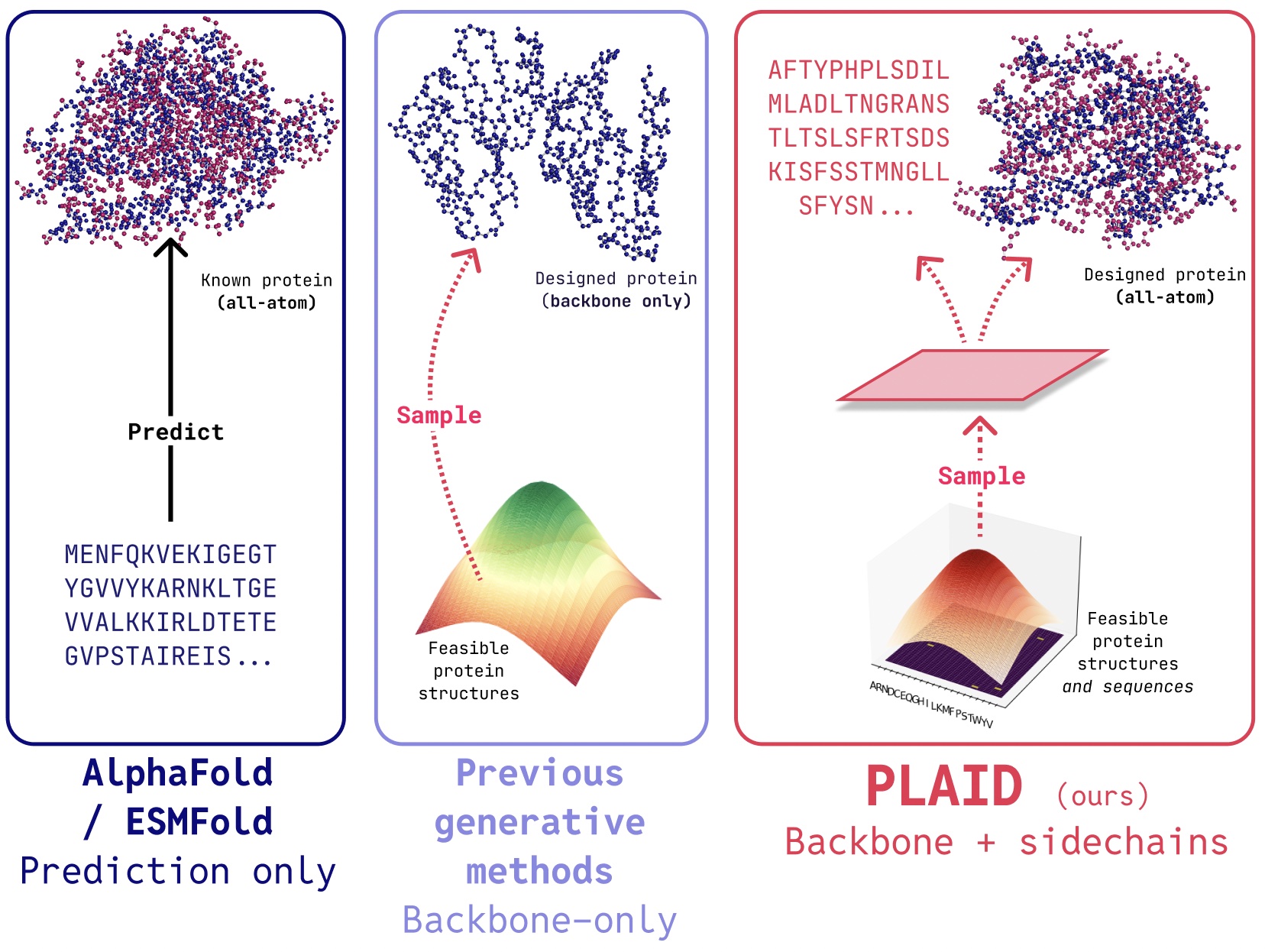
PLAID ist ein multimodales generatives Modell, das gleichzeitig Protein 1D -Sequenz und 3D -Struktur erzeugt, indem er den latenten Raum von Proteinfaltungsmodellen lernt.
Die Vergabe des 2024 Nobelpreis Alphafold2 markiert einen wichtigen Second der Anerkennung für die AI -Rolle in der Biologie. Was kommt als nächstes nach der Proteinfaltung?
In PLAIDWir entwickeln eine Methode, die lernt, aus dem latenten Raum der Proteinfaltungsmodelle zu Stichproben zu probieren erzeugen Neue Proteine. Es kann akzeptieren Kompositionsfunktion und Organismusaufforderungenund kann sein auf Sequenzdatenbanken trainiertdie 2-4 Größenordnungen sind, die größer sind als Strukturdatenbanken. Im Gegensatz zu vielen früheren Proteinstruktur-Generativmodellen befasst sich FLAID mit der Einstellung der multimodalen Co-Generationsprobleme: gleichzeitig sowohl diskrete Sequenz als auch kontinuierliche Strukturkoordinaten für All-Atom-Struktur erzeugen.
Von der Strukturvorhersage bis zum realen Arzneimitteldesign
Obwohl jüngste Arbeiten vielversprechend für die Fähigkeit von Diffusionsmodellen zeigen, Proteine zu erzeugen, gibt es immer noch Einschränkungen früherer Modelle, die sie für reale Anwendungen unpraktisch machen, wie z. B.:
- All-Atom-Technology: Viele vorhandene generative Modelle produzieren nur die Rückgratatome. Um die All-Atom-Struktur zu erzeugen und die Sidechain-Atome zu platzieren, müssen wir die Sequenz kennen. Dies schafft ein multimodales Erzeugungsproblem, das die gleichzeitige Erzeugung diskreter und kontinuierlicher Modalitäten erfordert.
- Organismusspezifität: Proteine Biologics, die für den menschlichen Gebrauch bestimmt sind humanisiertum nicht durch das menschliche Immunsystem zerstört zu werden.
- Kontrollspezifikation: Die Entdeckung von Arzneimitteln und die Inszenierung von Patienten ist ein komplexer Prozess. Wie können wir diese komplexen Einschränkungen angeben? Zum Beispiel können Sie auch nach der Angänzung der Biologie entscheiden, dass Tablets leichter zu transportieren sind als Fläschchen, was eine neue Einschränkung für die Löslichkeit hinzufügt.
Erzeugen von „nützlichen“ Proteinen
Das einfache Erzeugen von Proteinen ist nicht so nützlich wie Kontrolle die Technology zu bekommen nützlich Proteine. Wie könnte eine Schnittstelle für diesen Aussehen aussehen?

Betrachten wir zur Inspiration, wie wir die Bilderzeugung über kompositorische Textaufforderungen kontrollieren (Beispiel aus Liu et al., 2022).
In Plaid spiegeln wir diese Schnittstelle für wider Kontrollspezifikation. Das ultimative Ziel ist es, die Erzeugung vollständig über eine Textschnittstelle zu kontrollieren, aber hier betrachten wir die Kompositionsbeschränkungen für zwei Achsen als einen Proof-of-Idea: Funktion Und Organismus:
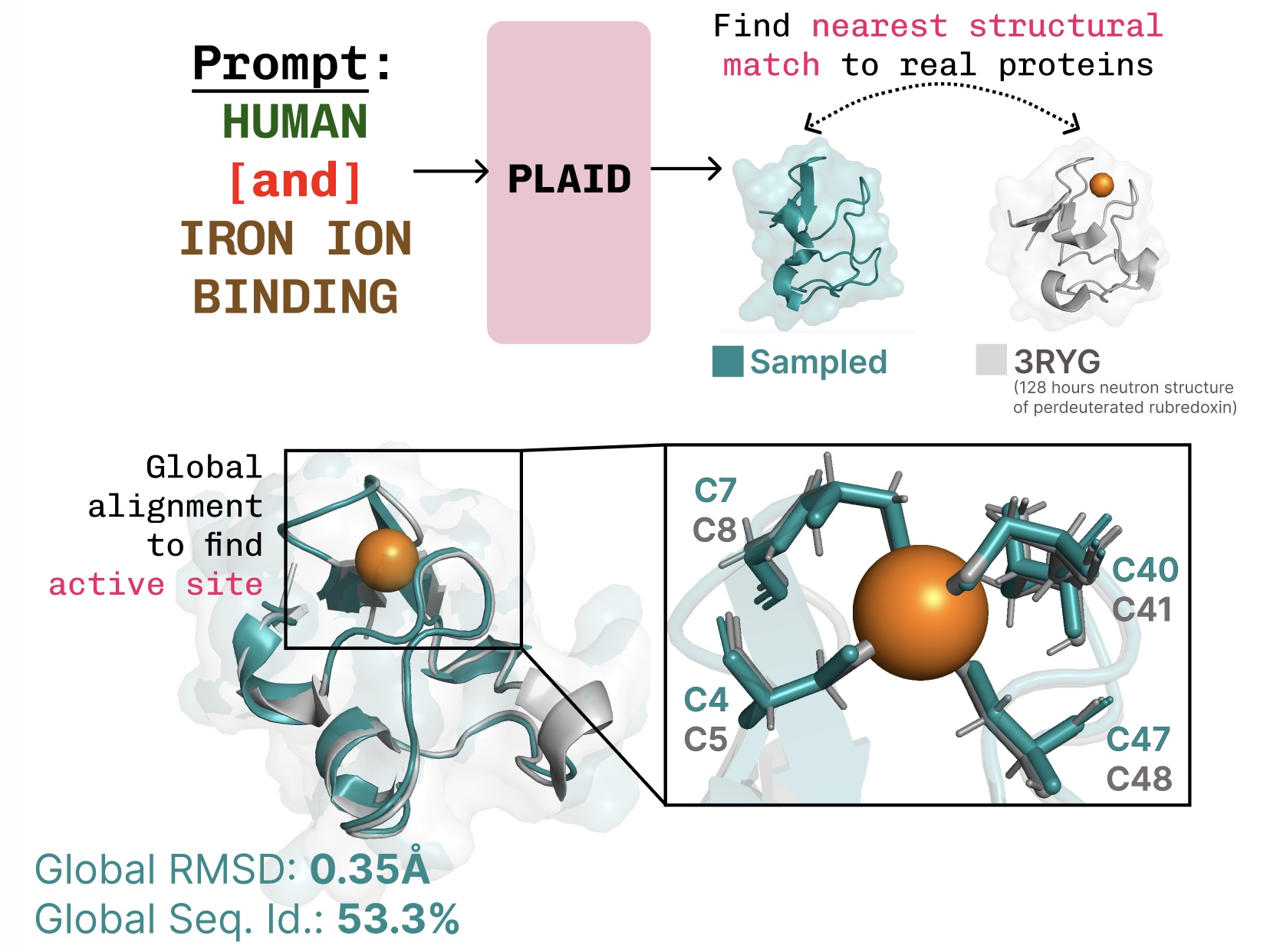
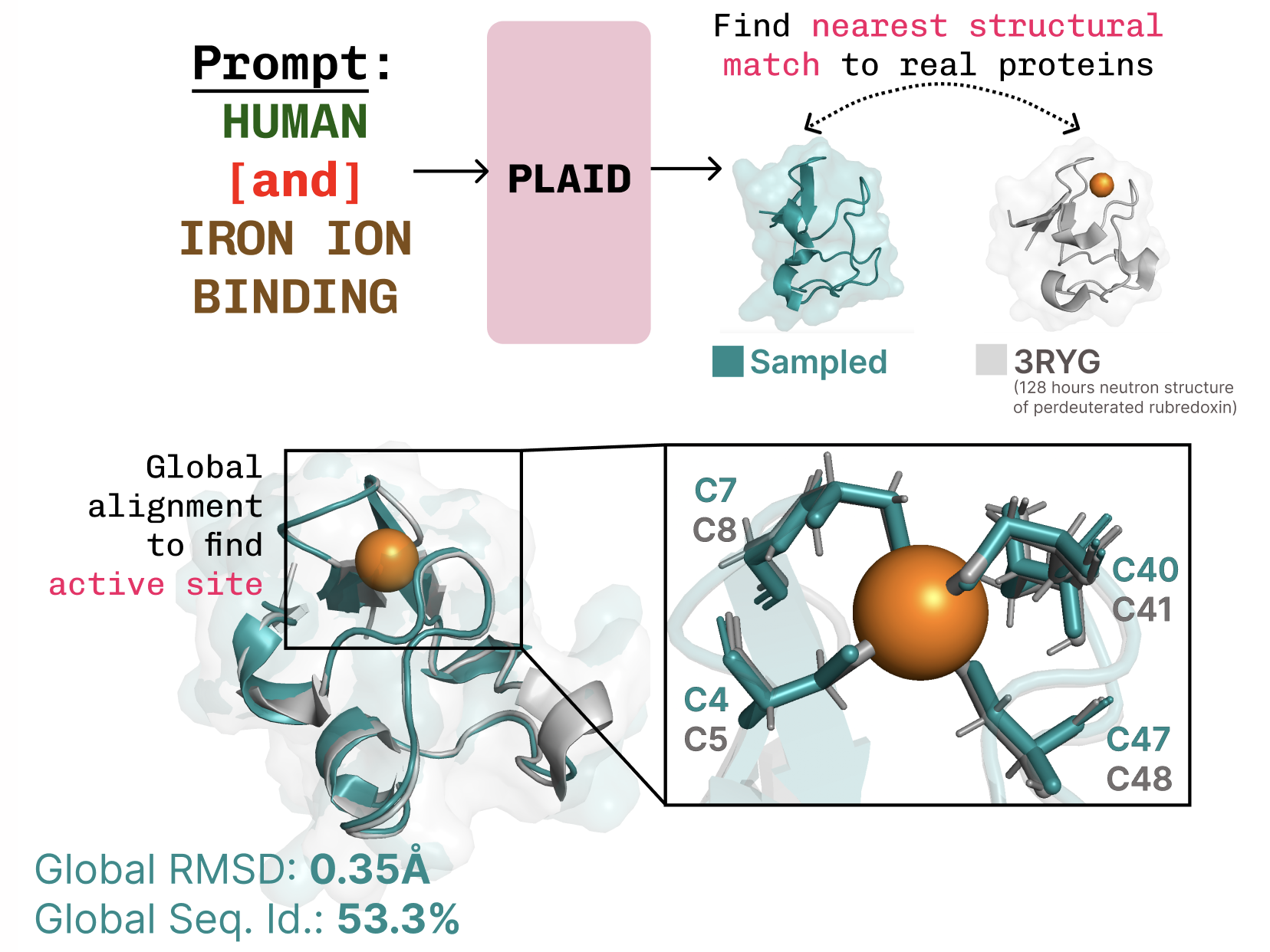
Erlernen der Funktionsstruktursequenzverbindung. Plaid lernt die tetraedrische Cystein-Fe2+/Fe3+ Das Koordinationsmuster, das häufig in Metalloproteinen zu finden ist, und gleichzeitig eine hohe Vielfalt auf Sequenzebene aufrechterhalten.
Coaching unter Verwendung von Trainingsdaten nur Sequenz
Ein weiterer wichtiger Aspekt des Plaidmodells ist, dass wir nur Sequenzen benötigen, um das generative Modell zu trainieren! Generative Modelle lernen die durch ihre Trainingsdaten definierte Datenverteilung und Sequenzdatenbanken sind erheblich größer als strukturelle, da Sequenzen viel billiger sind als die experimentelle Struktur.
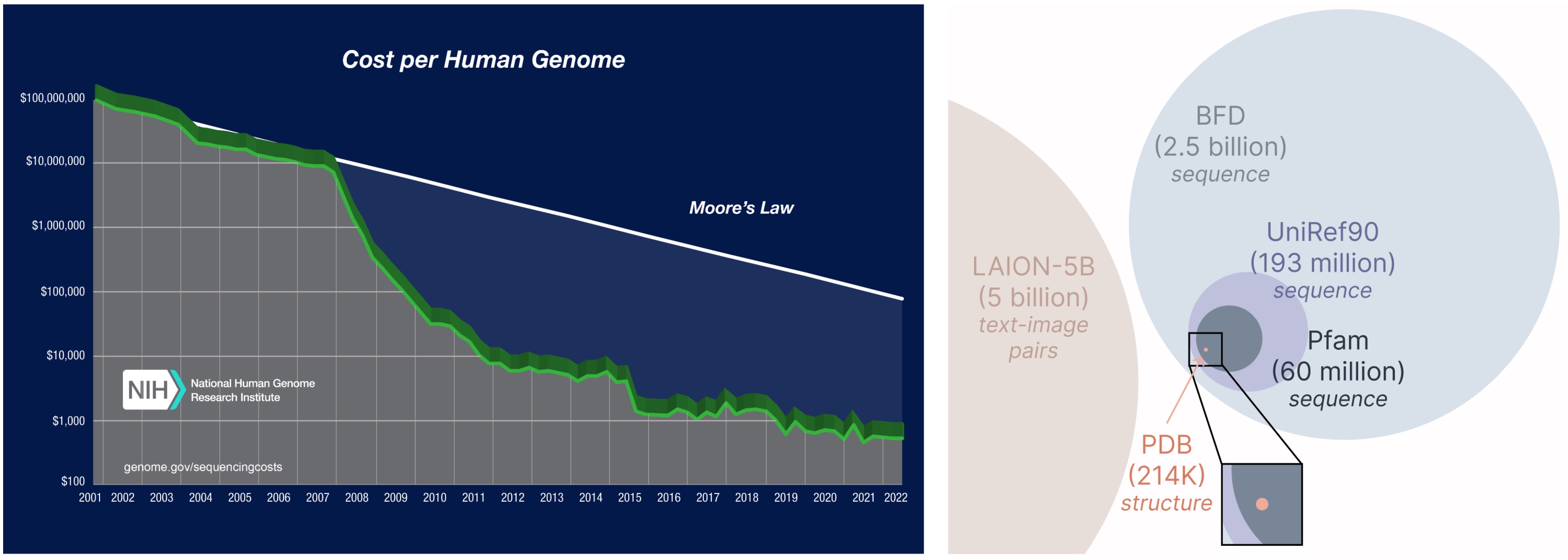
Lernen aus einer größeren und breiteren Datenbank. Die Kosten für die Erlangung von Proteinsequenzen sind viel niedriger als die experimentell charakterisierende Struktur, und Sequenzdatenbanken sind 2-4 Größenordnungen größer als strukturelle.
Wie funktioniert es?
Der Grund, warum wir das generative Modell trainieren können, um Struktur zu erzeugen, indem wir nur Sequenzdaten verwenden, ist das Erlernen eines Diffusionsmodells über dem latenter Raum eines Proteinfaltungsmodells. Dann können wir während der Inferenz nach der Probenahme aus diesem latenten Raum gültiger Proteine nehmen gefrorene Gewichte Aus dem Proteinfaltungsmodell, um die Struktur zu dekodieren. Hier verwenden wir Esmfoldein Nachfolger des Alphafold2 -Modells, das einen Abrufschritt durch ein Proteinsprachmodell ersetzt.
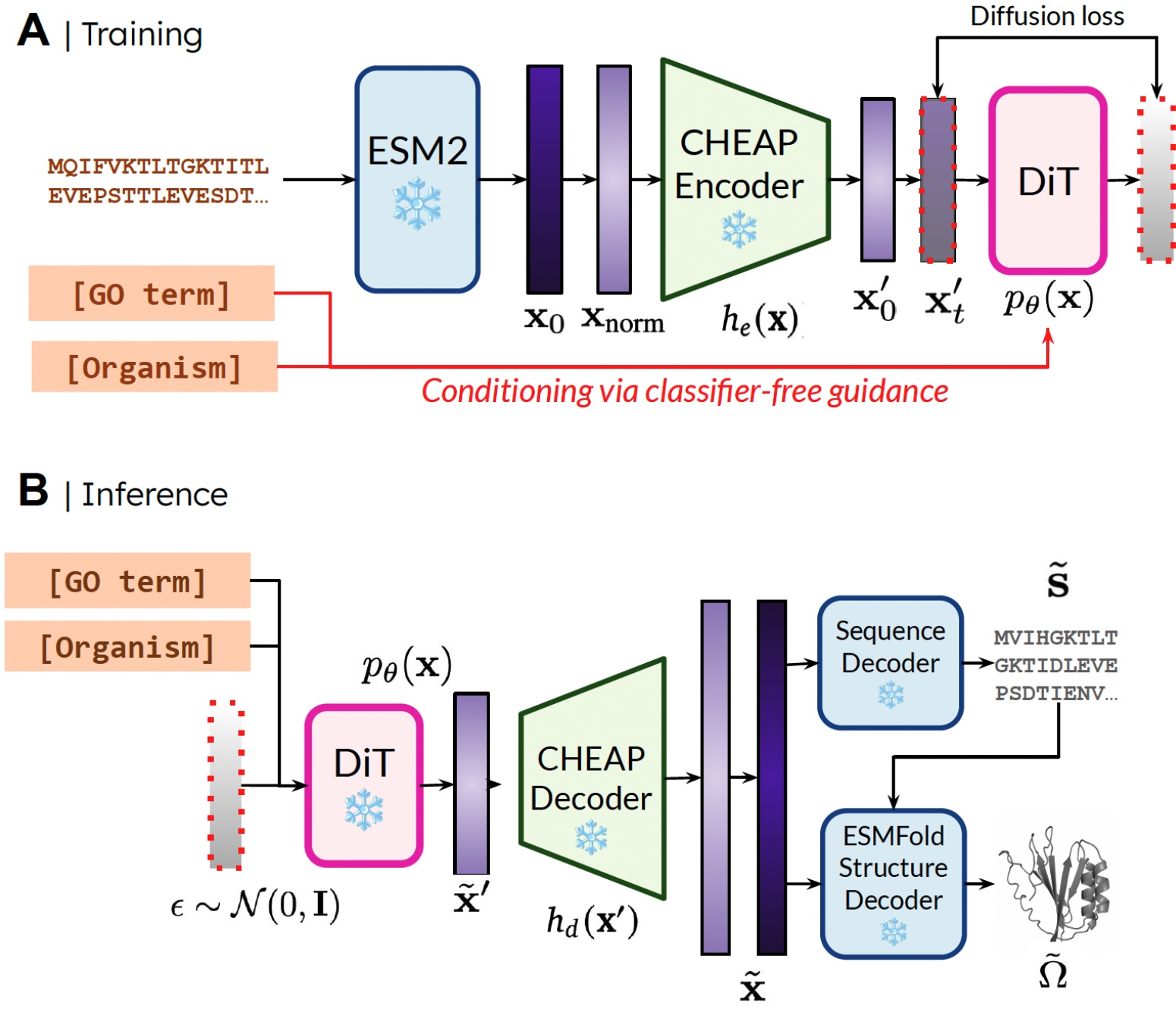
Unsere Methode. Während des Trainings sind nur Sequenzen erforderlich, um die Einbettung zu erhalten. Während der Inferenz können wir Sequenz und Struktur aus der Stichprobeneinbettung dekodieren. ❄️ bezeichnet gefrorene Gewichte.
Auf diese Weise können wir strukturelle Verständnisinformationen in den Gewichten von vorbereiteten Proteinfaltungsmodellen für die Proteindesign -Aufgabe verwenden. Dies ist analog zu, wie VLA-Modelle (Imaginative and prescient-Language-Motion) in Robotik Priors verwenden, die in Sehvermögensmodellen (VLMs) enthalten sind, die auf Daten im Internetmaßstab geschult sind, um die Wahrnehmung und das Argumentieren zu liefern und Informationen zu verstehen.
Komprimierung des latenten Raums der Proteinfaltungsmodelle
Eine kleine Falte mit direktem Anwenden dieser Methode ist, dass der latente Raum von ESMfold-in der Tat der latente Raum vieler transformatorbasierter Modelle-viel Regularisierung erfordert. Dieser Raum ist auch sehr groß, sodass das Erlernen dieser Einbettung die hochauflösende Bildsynthese kartiert.
Um dies anzugehen, schlagen wir auch vor BILLIG (Druckeinbettungsanpassungen von Proteinen)wo wir ein Kompressionsmodell für die Gelenkeinbettung von Proteinsequenz und Struktur lernen.
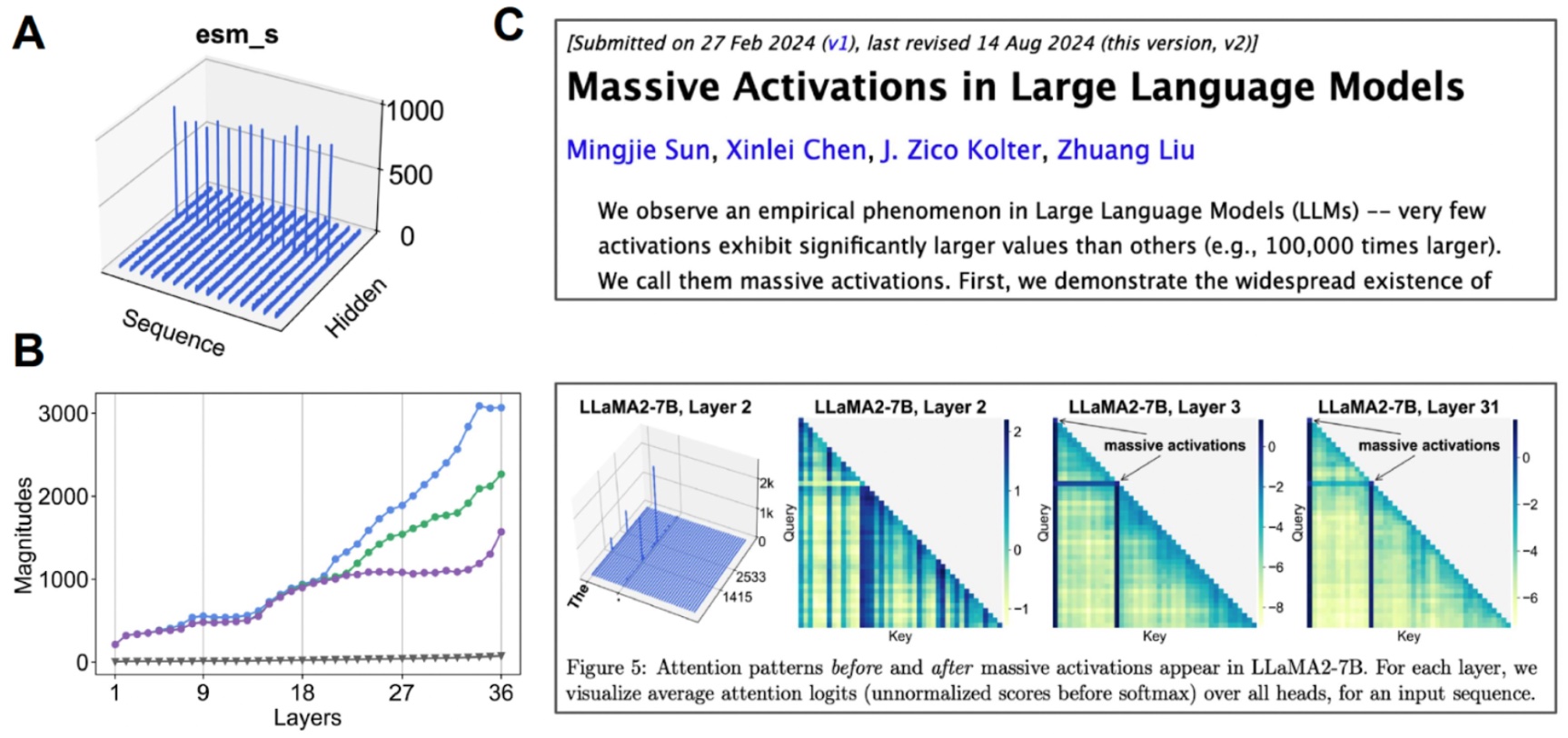
Untersuchung des latenten Raums. (A) Wenn wir den Mittelwert für jeden Kanal visualisieren, weisen einige Kanäle „huge Aktivierungen“ auf. (B) Wenn wir die Prime-3-Aktivierungen im Vergleich zum Medianwert (Grau) untersuchen, stellen wir fest, dass dies über viele Schichten passiert. (C) Für andere transformatorbasierte Modelle wurden auch huge Aktivierungen beobachtet.
Wir stellen fest, dass dieser latente Raum tatsächlich sehr komprimierbar ist. Durch ein bisschen mechanistische Interpretierbarkeit, um das Basismodell, mit dem wir arbeiten, besser zu verstehen, konnten wir ein All-Atom-Protein-Generativmodell erstellen.
Was kommt als nächstes?
Obwohl wir den Fall von Proteinsequenz und Strukturgenerierung in dieser Arbeit untersuchen, können wir diese Methode anpassen, um die multimodale Technology für alle Modalitäten durchzuführen, bei denen ein Prädiktor von einer reichhaltigeren Modalität zu einem weniger häufig vorkommenden. Da Sequenz-zu-Struktur-Prädiktoren für Proteine zunehmend komplexere Systeme in Angriff nehmen (EG Alphafold3 ist auch in der Lage, Proteine in Komplexen mit Nukleinsäuren und molekularen Liganden vorherzusagen), ist es leicht vorstellbar, dass die multimodale Erzeugung über komplexere Systeme unter Verwendung derselben Methode durchgeführt wird. Wenn Sie daran interessiert sind, zusammenzuarbeiten, um unsere Methode zu erweitern oder unsere Methode in der Moist-Lab zu testen, erreichen Sie bitte!
Weitere Hyperlinks
Wenn Sie unsere Artikel in Ihrer Forschung nützlich gefunden haben, sollten Sie die folgende Bibtex für Plaid und billig verwenden:
@article{lu2024generating,
title={Producing All-Atom Protein Construction from Sequence-Solely Coaching Knowledge},
creator={Lu, Amy X and Yan, Wilson and Robinson, Sarah A and Yang, Kevin Okay and Gligorijevic, Vladimir and Cho, Kyunghyun and Bonneau, Richard and Abbeel, Pieter and Frey, Nathan},
journal={bioRxiv},
pages={2024--12},
12 months={2024},
writer={Chilly Spring Harbor Laboratory}
}
@article{lu2024tokenized,
title={Tokenized and Steady Embedding Compressions of Protein Sequence and Construction},
creator={Lu, Amy X and Yan, Wilson and Yang, Kevin Okay and Gligorijevic, Vladimir and Cho, Kyunghyun and Abbeel, Pieter and Bonneau, Richard and Frey, Nathan},
journal={bioRxiv},
pages={2024--08},
12 months={2024},
writer={Chilly Spring Harbor Laboratory}
}
Sie können auch unsere Präprints überprüfen (PLAIDAnwesend BILLIG) und Codebasen (PLAIDAnwesend BILLIG).
Ein paar Spaß mit Bonusproteingeneration!
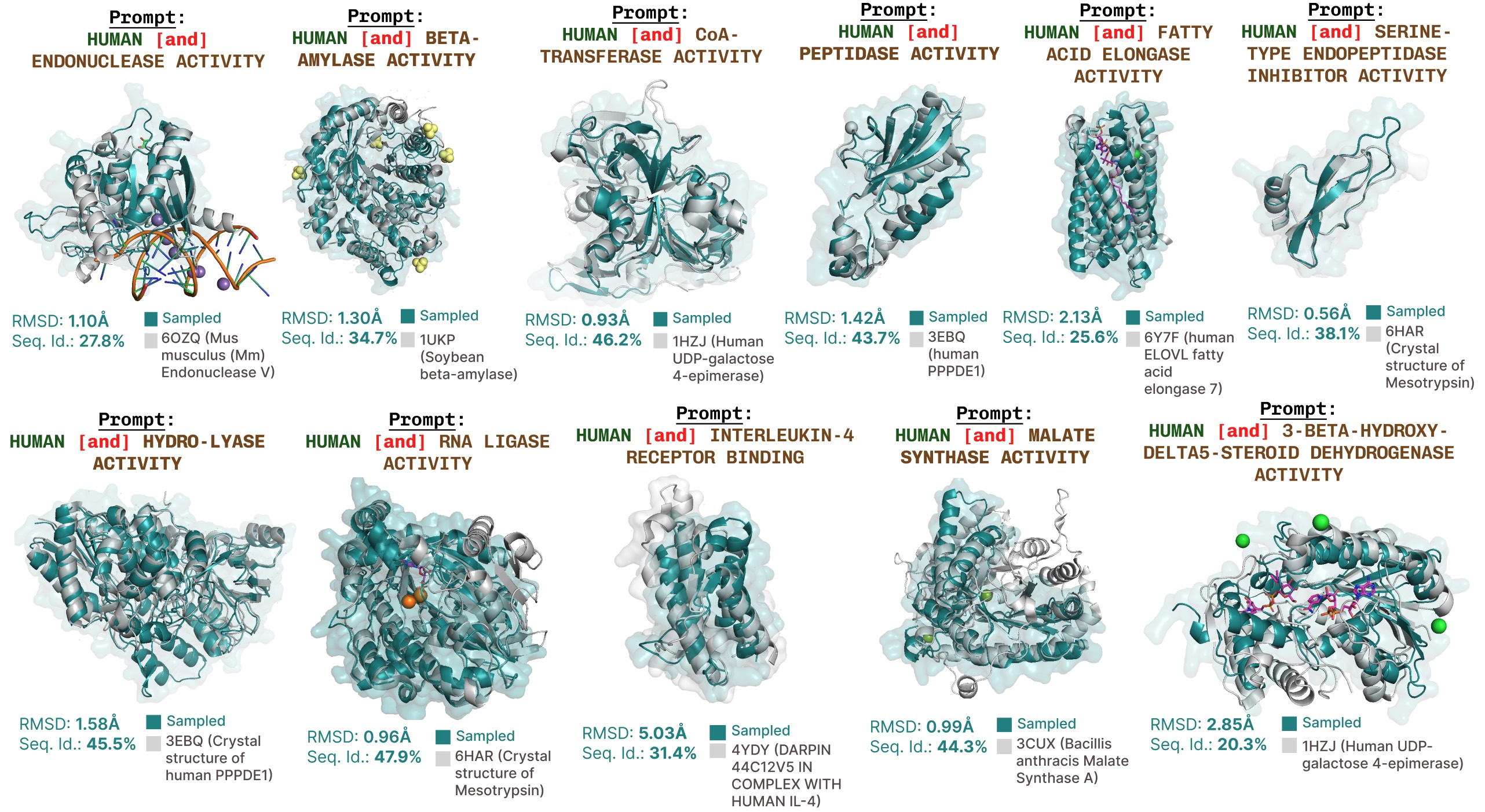
Zusätzliche funktionsgefüllte Generationen mit Plaid.
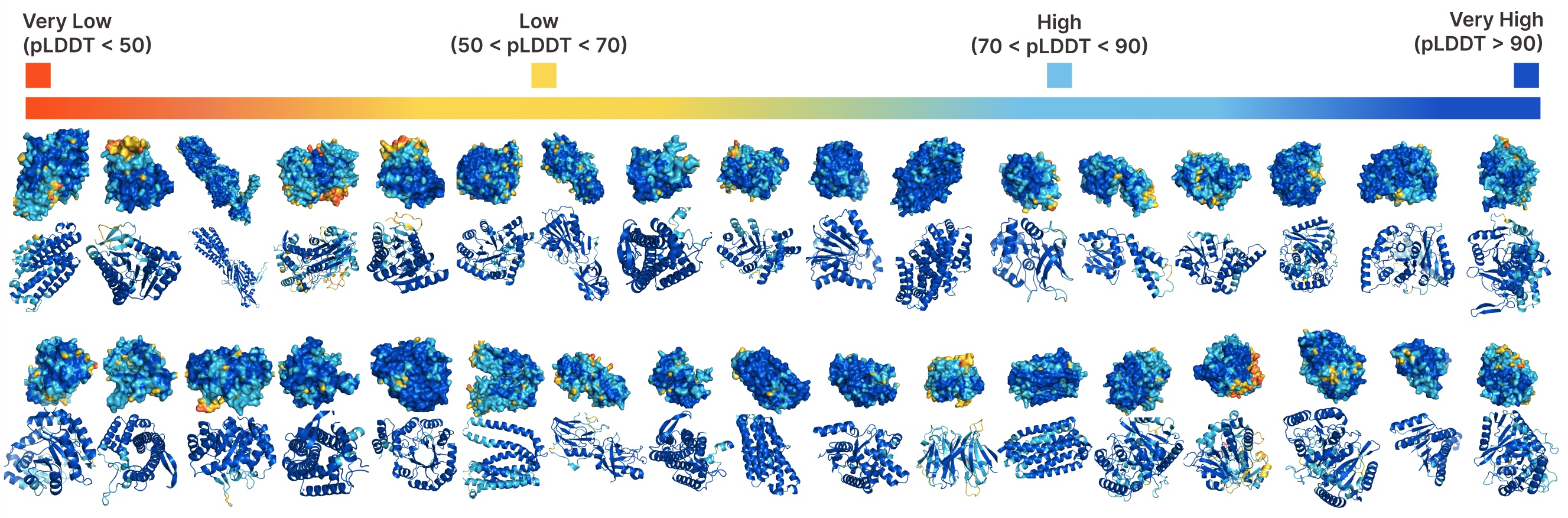
Bedingungslose Technology mit Plaid.
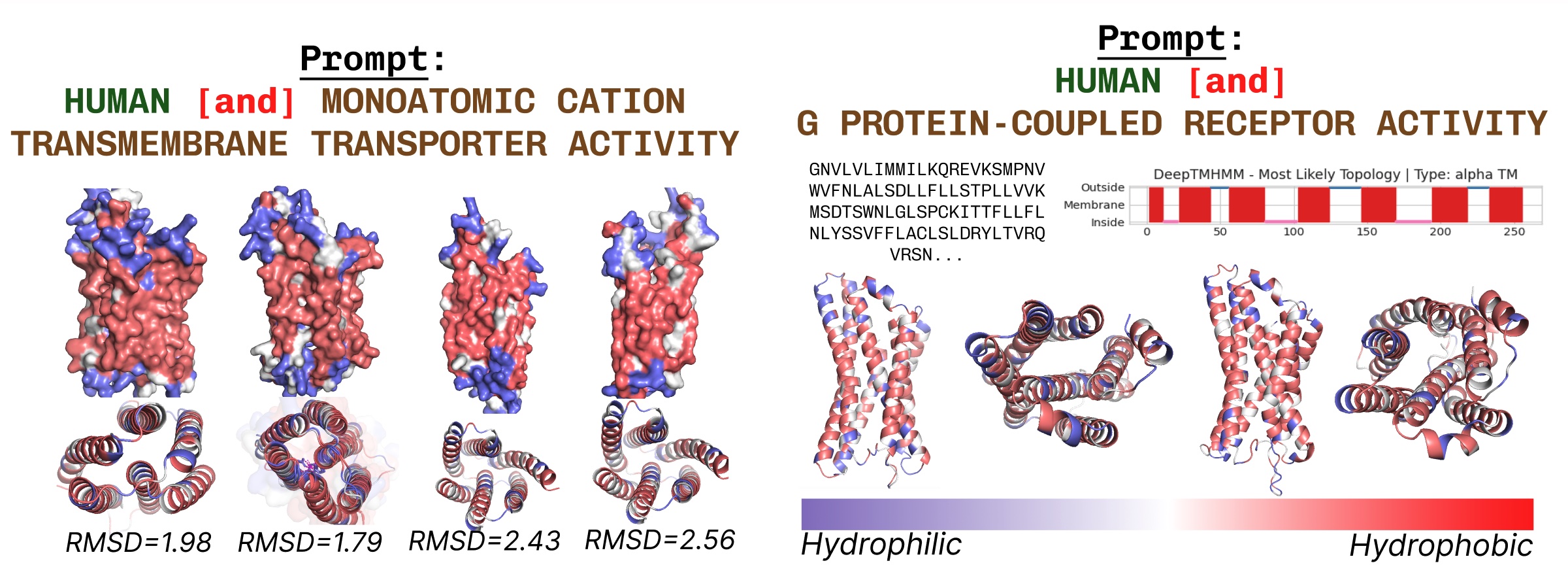
Transmembranproteine haben im Kern hydrophobe Reste, wo es in die Fettsäureschicht eingebettet ist. Diese werden konsistent beobachtet, wenn sie Plaid mit Transmembranprotein -Schlüsselwörtern beantragen.
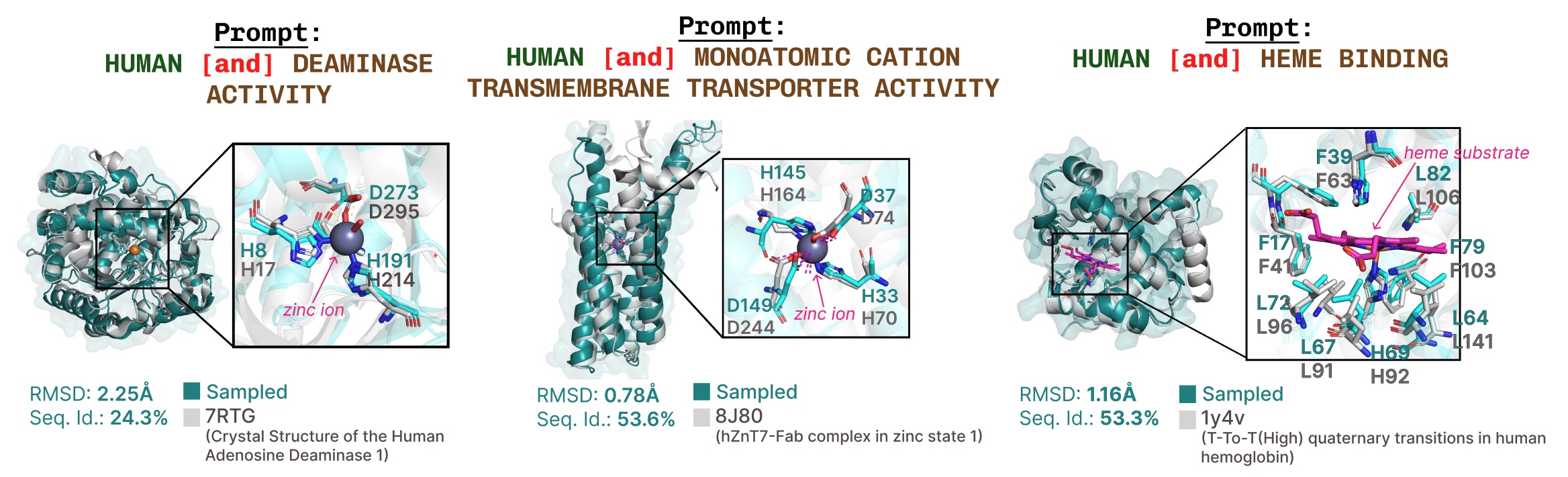
Zusätzliche Beispiele für die Rekapitulation der aktiven Stätte basierend auf der Aufforderung des Funktionsschlüsselworts.
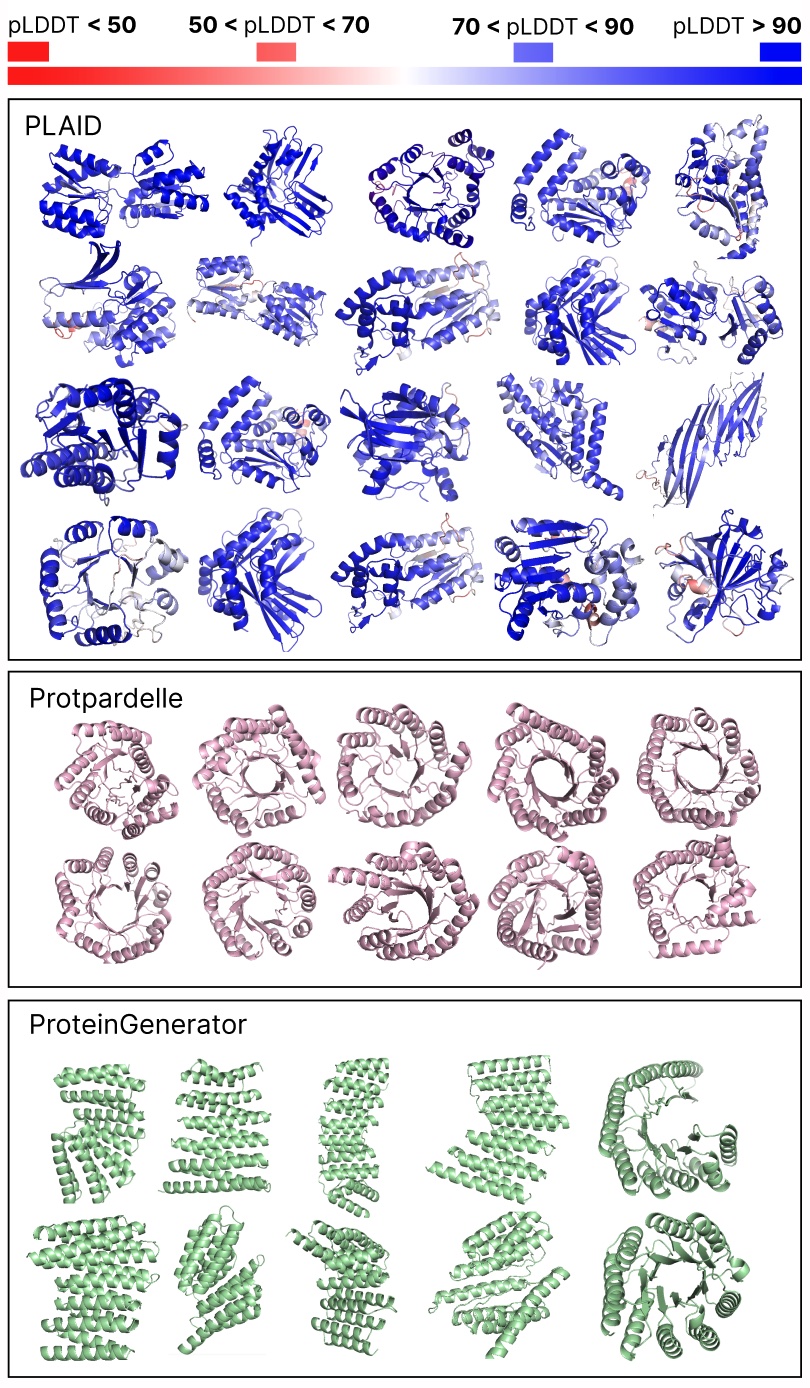
Vergleich von Proben zwischen Plaid- und All-Atom-Baselines. Plaid-Proben haben eine bessere Vielfalt und erfassen das Beta-Strang-Muster, das für Proteingenerative Modelle schwieriger zu lernen hat.
Anerkennung
Vielen Dank an Nathan Frey für ein detailliertes Suggestions zu diesem Artikel und an Co-Autoren in Bair, Genentech, Microsoft Analysis und der New York College: Wilson Yan, Sarah A. Robinson, Simon Kelow, Kevin Okay. Yang, Vladimir Gligorijevic, Kyunghyun Cho, Richard Bonneau, Pieter, Pieter und Nathan C.