
Proteine sind die Arbeitspferde, die unsere Zellen am Laufen halten, und es gibt viele tausend Proteine in unseren Zellen, die jeweils eine spezielle Funktion ausführen. Forscher wissen seit langem, dass die Struktur eines Proteins bestimmt, was es kann. In jüngerer Zeit werden Forscher zu schätzen wissen, dass die Lokalisierung eines Proteins auch für seine Funktion von entscheidender Bedeutung ist. Zellen sind voller Kompartimente, die dazu beitragen, ihre vielen Bewohner zu organisieren. Zusammen mit den bekannten Organellen, die die Seiten der Biologie-Lehrbücher schmücken, enthalten diese Räume auch eine Vielzahl dynamischer, membranloser Kompartimente, die bestimmte Moleküle zusammen konzentrieren, um gemeinsame Funktionen auszuführen. Zu wissen, wo ein bestimmtes Protein lokalisiert und mit wem es sich zusammenlasiert hat, kann daher nützlich sein, um das Protein und seine Rolle in der gesunden oder erkrankten Zelle besser zu verstehen. Forscher haben jedoch keine systematische Möglichkeit, diese Informationen vorherzusagen.
In der Zwischenzeit wurde die Proteinstruktur für mehr als ein halbes Jahrhundert untersucht, das im Instrument Alphafold im künstlichen Intelligenz gipfelte, das die Proteinstruktur aus dem Aminosäurecode eines Proteins vorhersagen kann, die lineare Schnur von Bausteinen darin, die sich falten, um seine Struktur zu erzeugen. Alphafold und Modelle wie es sind weit verbreitete Instruments in der Forschung geworden.
Proteine enthalten auch Regionen von Aminosäuren, die sich nicht in eine feste Struktur zusammenfalten, sondern auch wichtig für den Verbinden von Proteinen dynamischen Kompartimenten in der Zelle. MIT -Professor Richard Younger und Kollegen fragten sich, ob der Code in diesen Regionen zur Vorhersage der Proteinlokalisierung genauso verwendet werden könnte, wie andere Regionen zur Vorhersage der Struktur verwendet werden. Andere Forscher haben einige Proteinsequenzen entdeckt, die für die Proteinlokalisierung kodieren, und einige haben damit begonnen, Prädiktivmodelle für die Proteinlokalisierung zu entwickeln. Die Forscher wussten jedoch nicht, ob die Lokalisierung eines Proteins zu einem dynamischen Kompartiment auf der Grundlage seiner Sequenz vorhergesagt werden konnte, und sie hatten auch kein vergleichbares Werkzeug für Alphafold zur Vorhersage der Lokalisierung.
Jetzt jung, auch Mitglied des Whitehead Institute for Organic Analysis; Younger Lab Postdoc Henry Kilgore; Regina Barzilay, Faculty of Engineering Distinguished Professor für KI und Gesundheit im Abteilung für Elektrotechnik und Informatik sowie der Hauptuntersucher des MIT im Labor für Informatik und künstliche Intelligenz (CSAIL); und Kollegen haben ein solches Modell aufgebaut, das sie protGPS bezeichnen. In einem Papier, das auf veröffentlicht wurde 6. Februar im Journal WissenschaftMit den ersten Autoren Kilgore und Barzilay Lab-Studenten Itamar Chinn, Peter Mikhael und Ilan Mitnikov debütiert das interdisziplinäre Crew ihr Modell. Die Forscher zeigen, dass protGPs vorhersagen können, welcher von 12 bekannten Kompartimenten ein Protein lokalisiert wird, und ob eine krankheitsassoziierte Mutation diese Lokalisierung verändert. Darüber hinaus entwickelte das Forschungsteam einen generativen Algorithmus, der neuartige Proteine entwerfen kann, um in bestimmten Kompartimenten zu lokalisieren.
„Ich hoffe, dass dies ein erster Schritt in Richtung einer leistungsstarken Plattform ist, die es Menschen ermöglicht, Proteine zu studieren, um ihre Forschung zu machen“ Natürliche Prozesse und wie man therapeutische Hypothesen erzeugt und Medikamente zur Behandlung von Dysfunktionen in einer Zelle entwirft. “
Die Forscher validierten auch viele Vorhersagen des Modells mit experimentellen Exams in Zellen.
„Es hat mich sehr begeistert, dass ich in der Lage bin, vom Computerdesign bis hin zu diesen Dingen im Labor zu probieren“, sagt Barzilay. „In diesem Bereich von KI gibt es viele aufregende Papiere, aber 99,9 Prozent derjenigen werden nie in realen Systemen getestet. Dank unserer Zusammenarbeit mit dem jungen Labor konnten wir testen und wirklich lernen, wie intestine unser Algorithmus abschneidet. “
Entwicklung des Modells
Die Forscher bildeten ProtGPs auf zwei Chargen von Proteinen mit bekannten Lokalisierungen aus und testeten sie. Sie fanden heraus, dass es richtig vorhersagen konnte, wo Proteine eine hohe Genauigkeit haben. Die Forscher testeten auch, wie intestine protGPs Veränderungen der Proteinlokalisierung auf der Grundlage krankheitsassoziierter Mutationen innerhalb eines Proteins vorhersagen könnten. Es wurde festgestellt, dass viele Mutationen – Veränderungen der Sequenz für ein Gen und sein entsprechendes Protein – zur basierenden Assoziationsstudien beitragen oder zu Krankheiten führen, aber die Artwork und Weise, wie die Mutationen zu Krankheitssymptomen führen, bleiben unbekannt.
Es ist wichtig, den Mechanismus zu erfassen, wie eine Mutation zur Krankheit beiträgt, da Forscher Therapien entwickeln können, um diesen Mechanismus zu beheben und die Krankheit zu verhindern oder zu behandeln. Junge und Kollegen vermuteten, dass viele krankheitsassoziierte Mutationen durch Veränderung der Proteinlokalisierung zur Krankheit beitragen könnten. Zum Beispiel könnte eine Mutation dazu führen, dass ein Protein nicht in der Lage ist, einem Kompartiment mit wesentlichen Partnern zu beitreten.
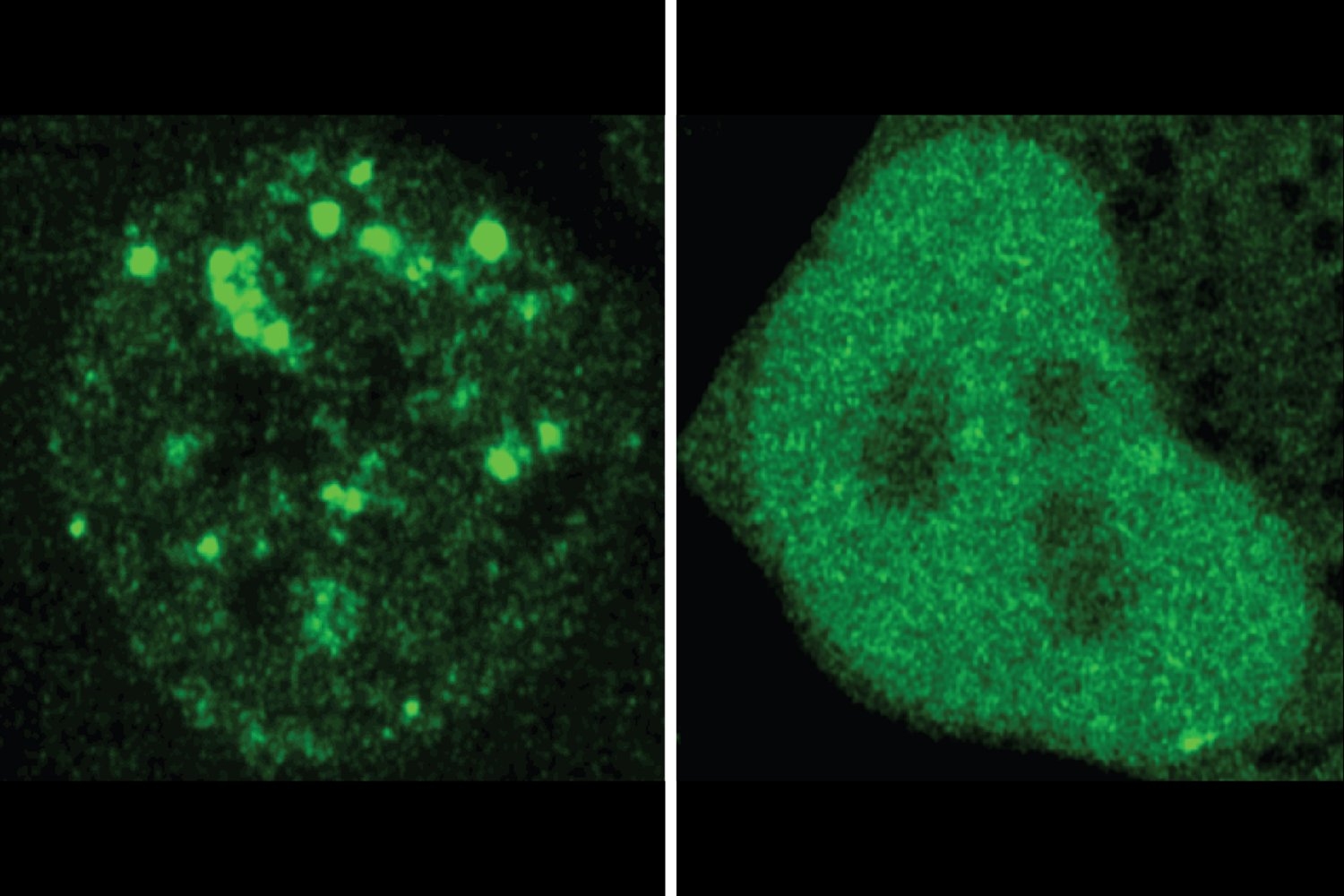
Sie testeten diese Hypothese, indem sie ProtgOs mehr als 200.000 Proteine mit krankheitsassoziierten Mutationen fütterten, und forderten sie dann auf, beide vorherzusagen, wo diese mutierten Proteine lokalisieren würden, wie stark ihre Vorhersage für ein gegebenes Protein von der Normalen zur mutierten Model verändert hat. Eine große Verschiebung der Vorhersage weist auf eine wahrscheinliche Veränderung der Lokalisierung hin.
Die Forscher fanden viele Fälle, in denen eine krankheitsassoziierte Mutation die Lokalisierung eines Proteins zu verändern schien. Sie testeten 20 Beispiele in Zellen und verwendeten Fluoreszenz, um zu vergleichen, wo in der Zelle ein normales Protein und die mutierte Model davon endeten. Die Experimente bestätigten die Vorhersagen von ProTGPS. Insgesamt stützen die Ergebnisse den Verdacht der Forscher, dass eine Fehl-Lokalisierung ein unterschätzter Krankheitsmechanismus sein kann und den Wert von ProTGPs als Instrument zum Verständnis von Krankheiten und zur Identifizierung neuer therapeutischer Wege demonstriert.
„Die Zelle ist ein so kompliziertes System mit so vielen Komponenten und komplexen Netzwerken von Interaktionen“, sagt Mitnikov. „Es ist tremendous interessant zu denken, dass wir mit diesem Ansatz das System beunruhigen, das Ergebnis davon sehen und so die Entdeckung von Mechanismen in der Zelle vorantreiben oder sogar Therapeutika entwickeln können.“
Die Forscher hoffen, dass andere ProgPs auf die gleiche Weise verwenden, wie sie prädiktive Strukturmodelle wie Alphafold verwenden und verschiedene Projekte zur Proteinfunktion, zur Dysfunktion und zur Krankheit vorantreiben.
Übergang über die Vorhersage der neuartigen Era hinausgehen
Die Forscher waren begeistert von den möglichen Verwendungen ihres Vorhersagemodells, aber sie wollten auch, dass ihr Modell über die Vorhersage von Lokalisationen vorhandener Proteine hinausgeht und ihnen ermöglicht, völlig neue Proteine zu entwerfen. Das Ziel struggle es, dass das Modell völlig neue Aminosäuresequenzen ausgeht, die sich an einem gewünschten Ort lokalisieren würden, wenn sie in einer Zelle gebildet wurden. Es ist unglaublich schwierig, ein neuartiges Protein zu erzeugen, das tatsächlich eine Funktion erfüllen kann – in diesem Fall – die Funktion der Lokalisierung eines bestimmten zellulären Kompartiments. Um die Erfolgschancen ihres Modells zu verbessern, haben die Forscher ihren Algorithmus nur so einen, dass Proteine wie die in der Natur zu findenen Entwerfen. Dies ist ein Ansatz, der aus logischen Gründen üblicherweise im Arzneimitteldesign verwendet wird. Die Natur hatte Milliarden von Jahren, um herauszufinden, welche Proteinsequenzen intestine funktionieren und welche nicht.
Aufgrund der Zusammenarbeit mit dem jungen Labor konnte das Crew für maschinelles Lernen testen, ob sein Proteingenerator arbeitete. Das Modell hatte gute Ergebnisse. In einer Runde erzeugte es 10 Proteine, die zum Nucleolus lokalisiert waren. Als die Forscher diese Proteine in der Zelle testeten, stellten sie fest, dass vier von ihnen im Nucleolus stark lokalisiert waren und andere möglicherweise auch leichte Verzerrungen zu diesem Ort hatten.
„Die Zusammenarbeit zwischen unseren Labors struggle für uns alle so generativ“, sagt Mikhael. „Wir haben gelernt, wie man die Sprachen des anderen spricht, in unserem Fall viel darüber gelernt, wie Zellen funktionieren, und durch die Möglichkeit, unser Modell experimentell zu testen, konnten wir herausfinden, was wir tun müssen, um tatsächlich zu machen Das Modell funktioniert und lässt es dann besser funktionieren. “
In der Lage zu sein, auf diese Weise funktionelle Proteine zu erzeugen, kann die Fähigkeit der Forscher verbessern, Therapien zu entwickeln. Wenn beispielsweise ein Medikament mit einem Ziel interagieren muss, das sich in einem bestimmten Fach lokalisiert, können Forscher dieses Modell verwenden, um ein Medikament dort zu entwerfen, um dort auch zu lokalisieren. Dies sollte das Arzneimittel wirksamer machen und die Nebenwirkungen verringern, da das Medikament mehr Zeit damit verbringen wird, sich mit seinem Ziel zu beschäftigen und weniger Zeit mit anderen Molekülen zu interagieren, was zu Auswirkungen von Off-Goal-Effekten führt.
Die Mitglieder des maschinellen Lernens sind begeistert von der Aussicht, das zu verwenden, was sie aus dieser Zusammenarbeit gelernt haben, um neuartige Proteine mit anderen Funktionen über die Lokalisierung hinaus zu entwerfen, die die Möglichkeiten für therapeutisches Design und andere Anwendungen erweitern würden.
„Viele Papiere zeigen, dass sie ein Protein entwerfen können, das in einer Zelle exprimiert werden kann, aber nicht, dass das Protein eine bestimmte Funktion hat“, sagt Chinn. „Wir hatten tatsächlich ein funktionelles Proteindesign und eine relativ große Erfolgsrate im Vergleich zu anderen generativen Modellen. Das ist wirklich aufregend für uns und etwas, auf das wir aufbauen möchten. “
Alle beteiligten Forscher sehen Progps als einen aufregenden Anfang an. Sie gehen davon aus, dass ihr Werkzeug verwendet wird, um mehr über die Rollen der Lokalisierung bei der Proteinfunktion und in der Fehllokalisierung bei Krankheiten zu erfahren. Darüber hinaus sind sie daran interessiert, die Lokalisierungsvorhersagen des Modells auf mehr Arten von Kompartimenten zu erweitern, mehr therapeutische Hypothesen zu testen und zunehmend funktionelle Proteine für Therapien oder andere Anwendungen zu entwickeln.
„Nachdem wir wissen, dass dieser Proteincode für die Lokalisierung vorhanden ist und dass maschinelle Lernmodelle diesen Code verstehen und sogar funktionelle Proteine mit seiner Logik erstellen können, die die Tür für so viele potenzielle Studien und Anwendungen öffnet“, sagt Kilgore.