
In den alten Zeiten – den wirklich alten Zeiten – struggle die Aufgabe, Materialien zu entwerfen, mühsam. Im Laufe von mehr als 1.000 Jahren versuchten Forscher, Gold herzustellen, indem sie Dinge wie Blei, Quecksilber und Schwefel in genau den richtigen Mischungsverhältnissen vermischten. Sogar berühmte Wissenschaftler wie Tycho Brahe, Robert Boyle und Isaac Newton versuchten sich an dem erfolglosen Unterfangen, das wir Alchemie nennen.
Die Materialwissenschaft hat natürlich einen langen Weg zurückgelegt. Seit 150 Jahren können Forscher auf das Periodensystem der Elemente zurückgreifen, das ihnen sagt, dass verschiedene Elemente unterschiedliche Eigenschaften haben und sich eines nicht auf magische Weise in ein anderes verwandeln kann. Darüber hinaus haben maschinelle Lernwerkzeuge in den letzten zehn Jahren unsere Fähigkeit, die Struktur und physikalischen Eigenschaften verschiedener Moleküle und Substanzen zu bestimmen, erheblich gesteigert. Neue Forschungen einer Gruppe unter der Leitung von Ju Li – Professor für Nukleartechnik am MIT und Professor für Materialwissenschaften und -technik der Tokyo Electrical Energy Firm – versprechen einen großen Sprung in den Fähigkeiten, die das Materialdesign erleichtern können. Die Ergebnisse ihrer Untersuchung werden in a berichtet Ausgabe Dezember 2024 von Naturinformatik.
Derzeit basieren die meisten maschinellen Lernmodelle, die zur Charakterisierung molekularer Systeme verwendet werden, auf der Dichtefunktionaltheorie (DFT), die einen quantenmechanischen Ansatz zur Bestimmung der Gesamtenergie eines Moleküls oder Kristalls durch Betrachtung der Elektronendichteverteilung bietet – das ist im Grunde die durchschnittliche Anzahl der Elektronen, die sich in einer Volumeneinheit um jeden gegebenen Punkt im Raum in der Nähe des Moleküls befinden. (Walter Kohn, der diese Theorie vor 60 Jahren miterfunden hat, erhielt dafür 1998 den Nobelpreis für Chemie.) Obwohl die Methode sehr erfolgreich struggle, weist sie laut Li einige Nachteile auf: „Erstens ist die Genauigkeit nicht gegeben durchweg großartig. Und zweitens sagt es nur eines: die niedrigste Gesamtenergie des molekularen Programs.“
„Paartherapie“ hilft
Sein Staff setzt nun auf eine andere rechnergestützte Chemietechnik, die ebenfalls aus der Quantenmechanik abgeleitet ist und als Coupled-Cluster-Theorie oder CCSD(T) bekannt ist. „Das ist der Goldstandard der Quantenchemie“, kommentiert Li. Die Ergebnisse von CCSD(T)-Berechnungen sind viel genauer als die Ergebnisse von DFT-Berechnungen und können ebenso vertrauenswürdig sein wie die Ergebnisse, die derzeit aus Experimenten gewonnen werden können. Das Drawback sei, dass die Durchführung dieser Berechnungen am Pc sehr langsam sei, sagt er, „und die Skalierung schlecht ist: Wenn man die Anzahl der Elektronen im System verdoppelt, werden die Berechnungen 100-mal teurer.“ Aus diesem Grund sind CCSD(T)-Berechnungen normalerweise auf Moleküle mit einer geringen Anzahl von Atomen beschränkt – in der Größenordnung von etwa 10. Alles, was darüber hinausgeht, würde einfach zu lange dauern.
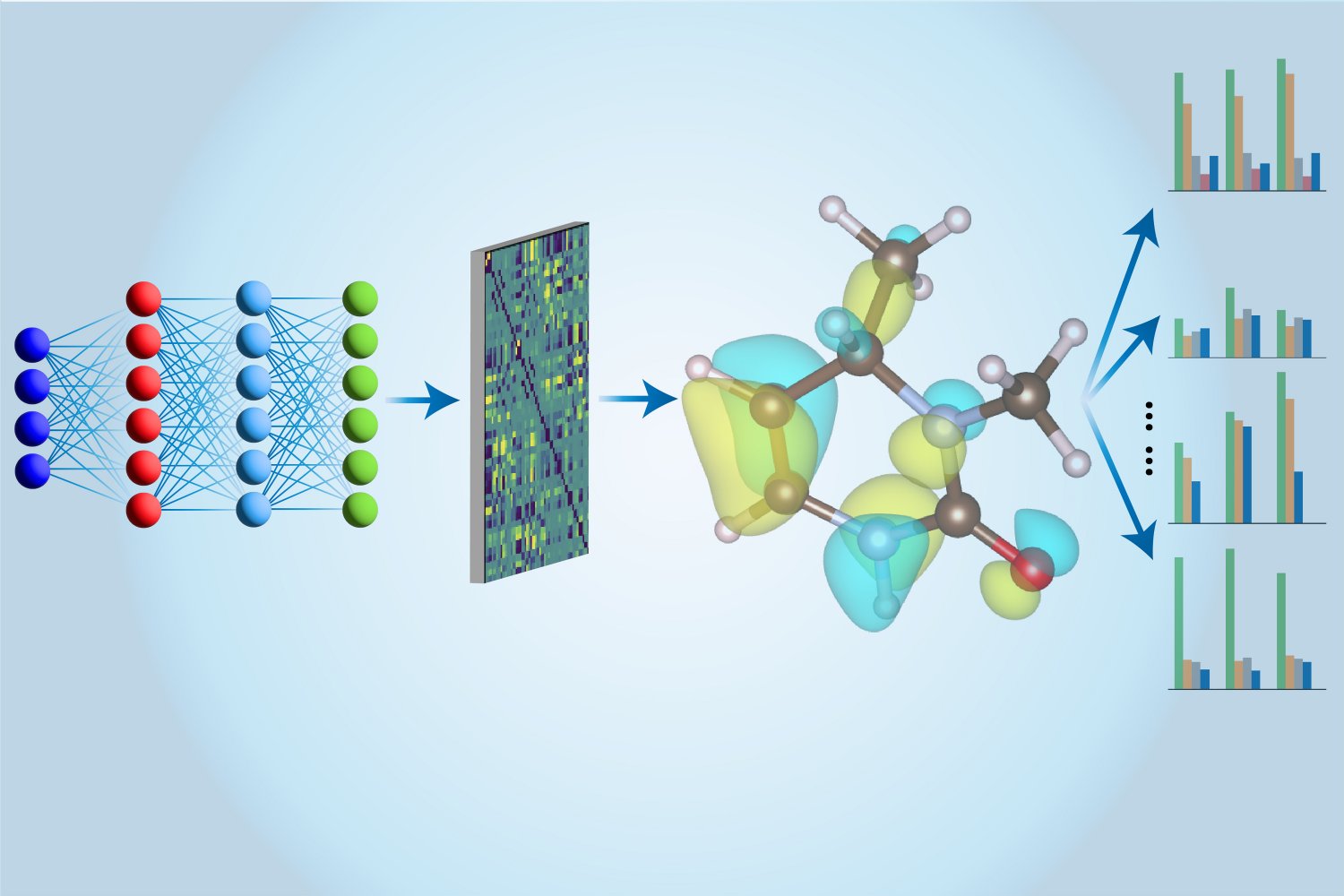
Hier kommt maschinelles Lernen ins Spiel. CCSD(T)-Berechnungen werden zunächst auf herkömmlichen Computern durchgeführt und die Ergebnisse dann zum Trainieren eines neuronalen Netzwerks mit einer neuartigen Architektur verwendet, die speziell von Li und seinen Kollegen entwickelt wurde. Nach dem Coaching kann das neuronale Netzwerk dieselben Berechnungen viel schneller durchführen, indem es Näherungstechniken nutzt. Darüber hinaus kann ihr neuronales Netzwerkmodell viel mehr Informationen über ein Molekül extrahieren als nur seine Energie. „In früheren Arbeiten haben Menschen mehrere verschiedene Modelle verwendet, um unterschiedliche Eigenschaften zu bewerten“, sagt Hao Tang, ein MIT-Doktorand in Materialwissenschaften und -technik. „Hier verwenden wir nur ein Modell, um alle diese Eigenschaften zu bewerten, weshalb wir es einen ‚Multi-Activity‘-Ansatz nennen.“
Das „Multi-Activity Digital Hamiltonian Community“ oder MEHnet gibt Aufschluss über eine Reihe elektronischer Eigenschaften, wie z. B. die Dipol- und Quadrupolmomente, die elektronische Polarisierbarkeit und die optische Anregungslücke – die Energiemenge, die benötigt wird, um ein Elektron aus dem zu nehmen Grundzustand in den niedrigsten angeregten Zustand. „Die Anregungslücke beeinflusst die optischen Eigenschaften von Materialien“, erklärt Tang, „weil sie die Frequenz des Lichts bestimmt, die von einem Molekül absorbiert werden kann.“ Ein weiterer Vorteil ihres CCSD-trainierten Modells besteht darin, dass es nicht nur Eigenschaften von Grundzuständen, sondern auch von angeregten Zuständen aufdecken kann. Das Modell kann auch das Infrarotabsorptionsspektrum eines Moleküls im Zusammenhang mit seinen Schwingungseigenschaften vorhersagen, wobei die Schwingungen der Atome innerhalb eines Moleküls miteinander gekoppelt sind, was zu verschiedenen kollektiven Verhaltensweisen führt.
Die Stärke ihres Ansatzes ist zu einem großen Teil auf die Netzwerkarchitektur zurückzuführen. Basierend auf der Arbeit des MIT-Assistenzprofessors Tess SmidtLaut Tang nutzt das Staff ein sogenanntes E(3)-äquivariantes graphisches neuronales Netzwerk, „in dem die Knoten Atome darstellen und die Kanten, die die Knoten verbinden, die Bindungen zwischen Atomen darstellen.“ Wir verwenden auch maßgeschneiderte Algorithmen, die physikalische Prinzipien – bezogen auf die Artwork und Weise, wie Menschen molekulare Eigenschaften in der Quantenmechanik berechnen – direkt in unser Modell integrieren.“
Testen, 1, 2 3
Bei der Analyse bekannter Kohlenwasserstoffmoleküle ergab das Modell von Li et al. übertrafen die DFT-Gegenstücke und stimmten weitgehend mit den experimentellen Ergebnissen aus der veröffentlichten Literatur überein.
Qiang Zhu – ein Materialentdeckungsspezialist an der College of North Carolina in Charlotte (der nicht an dieser Studie beteiligt struggle) – ist beeindruckt von dem, was bisher erreicht wurde. „Ihre Methode ermöglicht ein effektives Coaching mit einem kleinen Datensatz und erzielt gleichzeitig eine höhere Genauigkeit und Recheneffizienz im Vergleich zu bestehenden Modellen“, sagt er. „Dies ist eine spannende Arbeit, die die starke Synergie zwischen Computerchemie und Deep Studying veranschaulicht und neue Ideen für die Entwicklung genauerer und skalierbarer elektronischer Strukturmethoden bietet.“
Die am MIT ansässige Gruppe wandte ihr Modell zunächst auf kleine, nichtmetallische Elemente an – Wasserstoff, Kohlenstoff, Stickstoff, Sauerstoff und Fluor, aus denen organische Verbindungen hergestellt werden können – und ist seitdem dazu übergegangen, schwerere Elemente zu untersuchen: Silizium, Phosphor, Schwefel, Chlor und sogar Platin. Nachdem das Modell an kleinen Molekülen trainiert wurde, kann es auf immer größere Moleküle verallgemeinert werden. „Früher beschränkten sich die meisten Berechnungen auf die Analyse Hunderter Atome mit DFT und nur Dutzende Atome mit CCSD(T)-Berechnungen“, sagt Li. „Jetzt reden wir über den Umgang mit Tausenden von Atomen und am Ende vielleicht mit Zehntausenden.“
Derzeit werten die Forscher noch bekannte Moleküle aus, aber das Modell kann zur Charakterisierung bisher unbekannter Moleküle sowie zur Vorhersage der Eigenschaften hypothetischer Materialien verwendet werden, die aus verschiedenen Arten von Molekülen bestehen. „Die Idee besteht darin, mithilfe unserer theoretischen Werkzeuge vielversprechende Kandidaten auszuwählen, die bestimmte Kriterien erfüllen, und sie dann einem Experimentator zur Prüfung vorzuschlagen“, sagt Tang.
Es dreht sich alles um die Apps
Mit Blick auf die Zukunft ist Zhu optimistisch, was die möglichen Anwendungen angeht. „Dieser Ansatz birgt das Potenzial für ein molekulares Hochdurchsatz-Screening“, sagt er. „Das ist eine Aufgabe, bei der das Erreichen chemischer Genauigkeit von entscheidender Bedeutung für die Identifizierung neuartiger Moleküle und Materialien mit wünschenswerten Eigenschaften sein kann.“
Sobald sie die Fähigkeit beweisen, große Moleküle mit vielleicht Zehntausenden von Atomen zu analysieren, sollten wir laut Li in der Lage sein, neue Polymere oder Materialien zu erfinden, die bei der Entwicklung von Arzneimitteln oder in Halbleitergeräten verwendet werden könnten. Die Untersuchung schwererer Übergangsmetallelemente könnte zur Entwicklung neuer Materialien für Batterien führen – derzeit ein Bereich mit akutem Bedarf.
Die Zukunft, so sieht Li, ist weit offen. „Es geht nicht mehr nur um einen Bereich“, sagt er. „Letztendlich ist es unser Ziel, das gesamte Periodensystem mit einer Genauigkeit auf CCSD(T)-Niveau abzudecken, jedoch mit geringerem Rechenaufwand als DFT. Dies soll es uns ermöglichen, ein breites Spektrum an Problemen in der Chemie, Biologie und Materialwissenschaft zu lösen. Es ist derzeit schwer zu sagen, wie groß diese Spanne sein könnte.“
Diese Arbeit wurde vom Honda Analysis Institute unterstützt. Hao Tang dankt dem Mathworks Engineering Fellowship für seine Unterstützung. Die Berechnungen in dieser Arbeit wurden teilweise mit dem universellen Hochgeschwindigkeits-Atomsimulator Matlantis, dem Texas Superior Computing Heart, der MIT SuperCloud und dem Nationwide Power Analysis Scientific Computing durchgeführt.