Ein Protein im falschen Teil einer Zelle kann zu mehreren Krankheiten wie Alzheimer, Mukoviszidose und Krebs beitragen. In einer einzelnen menschlichen Zelle gibt es jedoch ungefähr 70.000 verschiedene Proteine und Proteinvarianten, und da Wissenschaftler normalerweise nur für eine Handvoll in einem Experiment testen können, ist es äußerst kostspielig und zeitaufwändig, die Standorte von Proteinen manuell zu identifizieren.
Eine neue Technology von Computertechniken versucht, den Prozess mithilfe von maschinellen Lernmodellen zu optimieren, die häufig Datensätze nutzen, die Tausende von Proteinen und deren Standorten enthalten, gemessen über mehrere Zelllinien. Einer der größten solcher Datensätze ist der menschliche Proteinatlas, der das subzelluläre Verhalten von Over katalogisiert 13.000 Proteine in mehr als 40 Zelllinien. So enorm es auch ist, der menschliche Proteinatlas hat nur etwa 0,25 Prozent aller möglichen Paarungen aller Proteine und Zelllinien innerhalb der Datenbank untersucht.
Jetzt haben Forscher der MIT, der Harvard College und des Broad Institute of MIT und Harvard einen neuen rechnerischen Ansatz entwickelt, der den verbleibenden unbekannten Raum effizient untersuchen kann. Ihre Methode kann die Place eines Proteins in einer menschlichen Zelllinie vorhersagen, auch wenn sowohl Protein als auch Zelle noch nie zuvor getestet wurden.
Ihre Technik geht einen Schritt weiter als bei vielen auf AI-basierten Methoden, indem ein Protein auf Einzelzellenebene lokalisiert wird, anstatt als gemittelte Schätzung über alle Zellen eines bestimmten Typs. Diese Einzelzelllokalisierung könnte beispielsweise nach der Behandlung die Place eines Proteins in einer bestimmten Krebszelle festlegen.
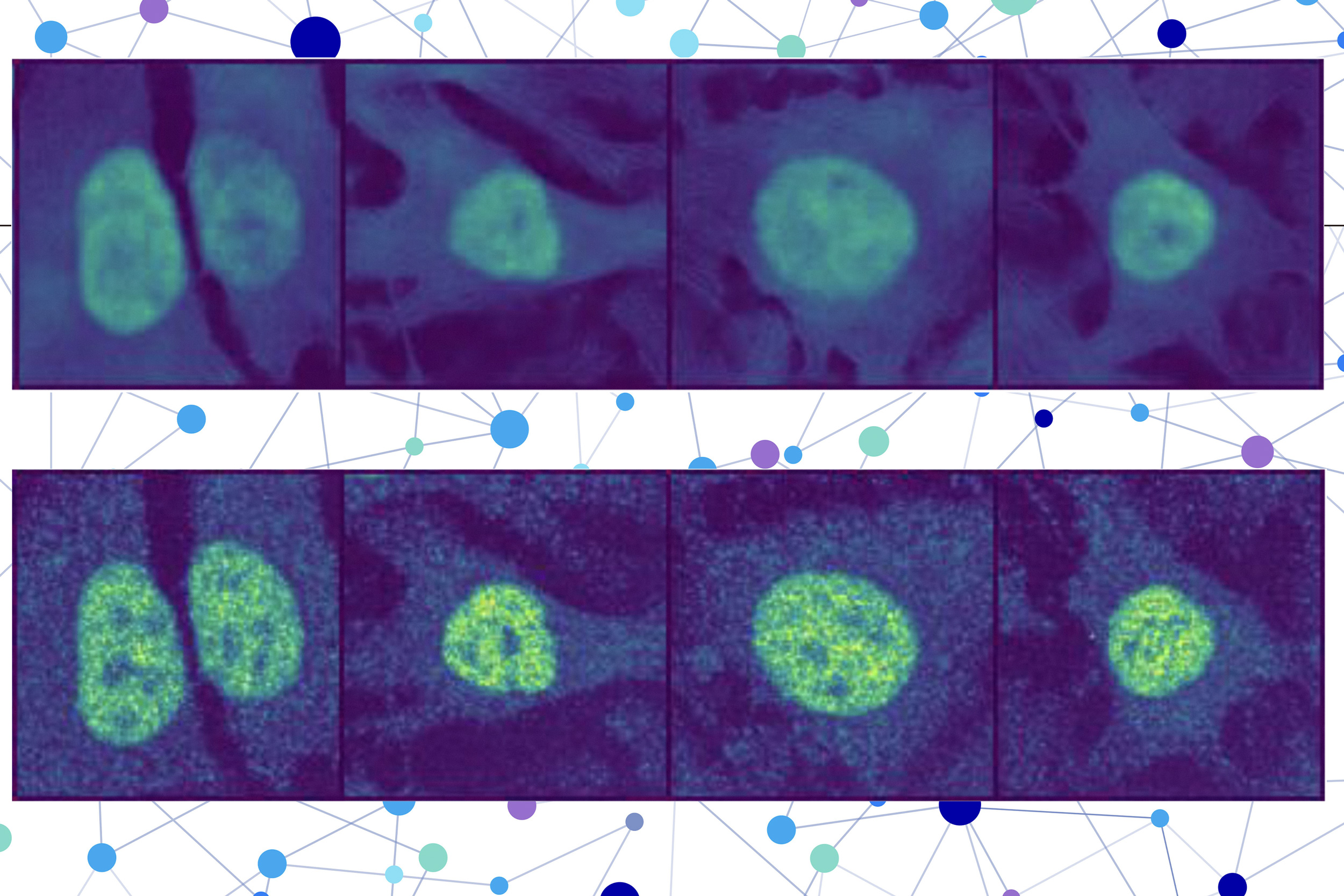
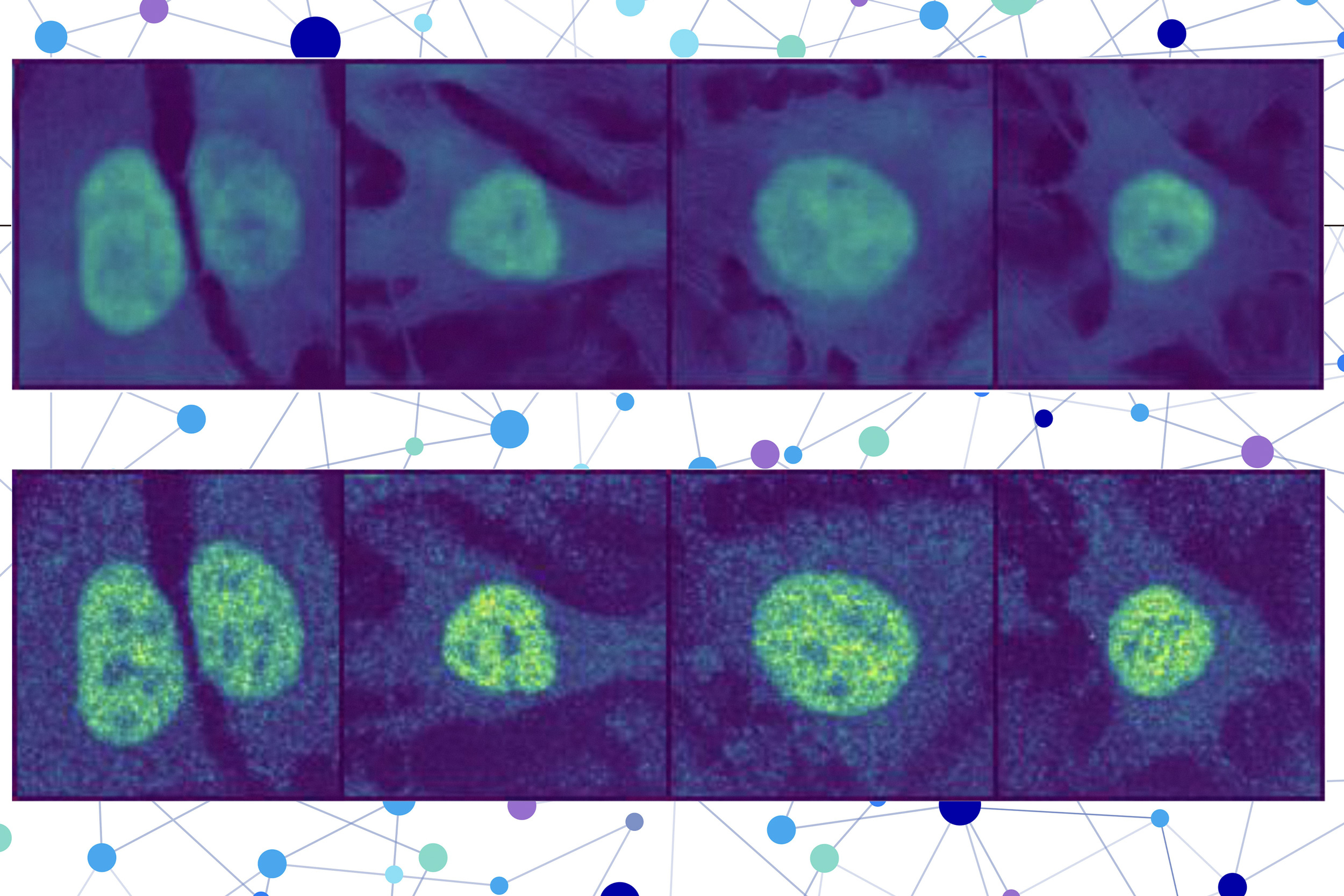
Die Forscher kombinierten ein Proteinsprachmodell mit einem speziellen Pc -Imaginative and prescient -Modell, um reichhaltige Particulars über Protein und Zelle zu erfassen. Am Ende erhält der Benutzer ein Bild einer Zelle mit einem hervorgehobenen Teil, der auf die Vorhersage des Modells hinweist, wo sich das Protein befindet. Da die Lokalisierung eines Proteins auf ihren Funktionsstatus hinweist, könnte diese Technik Forschern und Klinikern helfen, Krankheiten effizienter zu diagnostizieren oder Arzneimittelziele zu identifizieren und gleichzeitig Biologen zu verstehen, wie komplexe biologische Prozesse mit der Proteinlokalisierung zusammenhängen.
„Sie könnten diese Protein-Lokalisierungsexperimente auf einem Pc durchführen, ohne eine Laborbank zu berühren und sich hoffentlich monatelanges Mühe zu sparen. Während Sie die Vorhersage weiterhin überprüfen müssten, könnte diese Technik wie ein anfängliches Screening des Testens für experimentelles Testen wirken“, sagt Yitong Tseo, ein Abschluss in MIT-Programm- und Systembiologie-Programm- und Co-Lead-Autoren, auf diesen Forschungen.
TSEO wird vom Co-Lead-Autor Xinyi Zhang, einem Doktorand der Abteilung für Elektrotechnik und Informatik (EECS) und des Eric und Wendy Schmidt Middle des Broad Institute, auf dem Papier begleitet. Yunhao Bai vom Broad Institute; und hochrangige Autoren Fei Chen, Assistenzprofessorin bei Harvard und Mitglied des Broad Institute, und Caroline Uhler, Andrew und Erna Viterbi Professor für Ingenieurwesen in EECs und das MIT Institute for Knowledge, Methods und Society (IDSS), der auch Direktor der INIC- und Wendy -Schmidt -Zentrum sowie in den Labor- und Entscheidungssystemen ist. Die Forschung erscheint heute in Naturmethoden.
Zusammenarbeit Modelle
Viele vorhandene Proteinvorhersagemodelle können nur Vorhersagen auf der Grundlage der Protein- und Zelldaten treffen, auf denen sie trainiert wurden oder die Place eines Proteins innerhalb einer einzelnen Zelle nicht genau bestimmen können.
Um diese Einschränkungen zu überwinden, erstellten die Forscher eine zweiteilige Methode zur Vorhersage des subzellulären Standorts der unsichtbaren Proteine, die als Welpen bezeichnet werden.
Der erste Teil verwendet ein Proteinsequenzmodell, um die lokalisierungsbestimmenden Eigenschaften eines Proteins und seiner 3D-Struktur basierend auf der Kette von Aminosäuren zu erfassen.
Der zweite Teil enthält ein Picture -Inpainting -Modell, mit dem fehlende Teile eines Bildes ausgefüllt werden sollen. Dieses Pc -Imaginative and prescient -Modell befasst sich mit drei gefärbten Bildern einer Zelle, um Informationen über den Zustand dieser Zelle zu sammeln, wie z. B. ihre Artwork, individuelle Merkmale und ob es unter Stress steht.
Welpe verbinden sich den von jedem Modell erstellten Darstellungen, um vorherzusagen, wo sich das Protein in einer einzelnen Zelle befindet, und verwendet einen Bilddecoder, um ein hervorgehobenes Bild auszugeben, das den vorhergesagten Ort anzeigt.
„Verschiedene Zellen innerhalb einer Zelllinie zeigen unterschiedliche Eigenschaften, und unser Modell kann diese Nuance verstehen“, sagt Tseo.
Ein Benutzer gibt die Abfolge von Aminosäuren ein, die das Protein und drei Zellfärbungsbilder bilden – eine für den Kern, einen für die Mikrotubuli und eine für das endoplasmatische Retikulum. Dann macht Welpen den Relaxation.
Ein tieferes Verständnis
Die Forscher verwendeten während des Schulungsprozesses einige Tips, um Welpen beizubringen, wie sie Informationen aus jedem Modell so kombinieren können, dass es eine fundierte Vermutung über den Standort des Proteins erraten kann, auch wenn es dieses Protein noch nicht gesehen hat.
Zum Beispiel weisen sie dem Modell eine sekundäre Aufgabe während des Trainings zu: so explizit das Kompartiment der Lokalisierung wie den Zellkern zu benennen. Dies erfolgt neben der primären Inpainting -Aufgabe, um das Modell effektiver zu lernen.
Eine gute Analogie könnte ein Lehrer sein, der ihre Schüler auffordert, alle Teile einer Blume zu zeichnen und ihre Namen zu schreiben. Dieser zusätzliche Schritt wurde festgestellt, dass das Modell sein allgemeines Verständnis der möglichen Zellkompartimente verbessert.
Darüber hinaus hilft die Tatsache, dass Welpen gleichzeitig auf Proteinen und Zelllinien trainiert werden, ein tieferes Verständnis dafür, wo in einem Zellbildproteine zu lokalisieren ist.
Welpen können sogar selbst verstehen, wie unterschiedliche Teile der Sequenz eines Proteins getrennt zu seiner Gesamtlokalisierung beitragen.
„Die meisten anderen Methoden erfordern normalerweise, dass Sie zuerst eine Färbung des Proteins haben. Sie haben es additionally bereits in Ihren Trainingsdaten gesehen. Unser Ansatz ist insofern einzigartig, als sie gleichzeitig Proteine und Zelllinien überallieren können“, sagt Zhang.
Da Welpen auf unsichtbare Proteine verallgemeinern können, kann sie Veränderungen der Lokalisierung erfassen, die durch einzigartige Proteinmutationen angetrieben werden, die nicht in den menschlichen Proteinatlas enthalten sind.
Die Forscher verifizierten, dass Welpen den subzellulären Ort neuer Proteine in unsichtbaren Zelllinien durch Durchführung von Laborversuche vorhersagen könnten und die Ergebnisse verglichen. Darüber hinaus zeigten die Welpen im Vergleich zu einer AI -Foundation -AI -Methode im Durchschnitt weniger Vorhersagemangel über die von ihnen getesteten Proteine.
In Zukunft möchten die Forscher die Welpen verbessern, damit das Modell Protein-Protein-Wechselwirkungen verstehen und Lokalisierungsvorhersagen für mehrere Proteine in einer Zelle vornehmen kann. Langfristig wollen sie es den Welpen ermöglichen, Vorhersagen in Bezug auf das lebende menschliche Gewebe und nicht in kultivierten Zellen zu treffen.
Diese Forschung wird vom Eric und Wendy Schmidt Middle des Broad Institute, den Nationwide Institutes of Well being, der Nationwide Science Basis, dem Burroughs Welcome Fund, der Searle Students Basis, dem Harvard Stem Cell Institute, dem Merkin -Institut, dem Amt für Marineforschung und dem Ministerium für Energie, finanziert.